I started working on what I suppose you could call Foundations of Mathematics when I was in Sweden back in 2019, but I’m reluctant to call it that, because I didn’t produce any meaningful axiomatic systems, or results, though it looks like I did solve the Continuum Hypothesis (but I didn’t realize it at the time). Because I was producing an incredible amount of useful work in Physics and Artificial Intelligence, I gave up my work on Foundations of Mathematics, picking it up here and there.
One problem that’s always bothered me is the affect Mathematics has on the observable world, that is plainly beyond Physics. Specifically, all combinatorial properties of a system (e.g., counting or connective properties) are always true, with certainty, for a given system, at all moments in time. For example, if a system has 5 components, then there are 5 choose 2 = 10 ways of selecting any pair of components from the system. This is never subject to serious doubt, and what’s even more strange, is that there’s no process that causes the Theorems of Combinatorics to hold. In more formal terms, the belief in the veracity of the Theorems of Combinatorics is not empirical, and its machinations (for lack of a better word), appear to be atemporal, and eternal, in the most real sense, that they do not begin or end, and instead simply apply at all times, at all points in space.
Observations of this sort in my opinion take what was previously Philosophy, and turn it into something that is close to Mathematics and Science, and I think I have my first real result in what could be an entirely new way of thinking about reality and Mathematics. Specifically, I believe I’ve constructed a credible argument that (i) Mathematics exists in 3-space, i.e., what we would call the observable world, and (ii) Mathematics does not have a location.
The argument is as follows:
Assumption 1: If a system does not exist, then that system cannot have an effect on the observable world.
Now assume that Mathematics does not exist. We just showed above that Mathematics certainly has an admittedly peculiar, but immutable effect on the observable world, and in fact, on all systems in the observable world. Therefore, mathematics exists.
I don’t want to over formalize prong (ii), but I think it’s fair to say there is no known cosmic ledger that contains all the true of Theorems of Mathematics. If you want to convince yourself that’s an empirical result, that’s fine by me, but in my life, I’m going to treat that as a given, and therefore, Mathematics exists in the observable world, but does not have a location in the observable world, and instead only has effects on the observable world.
Tonight, I derived what I think is a fascinating corollary, by analyzing and abstracting the structure of the argument, which is in essence a forbidden property. Specifically, if a system does not exist, then it cannot have property P, which in Assumption 1, is an observable effect. Now ask whether there is some other property P*, specifically:
Query 1: Is there some other forbidden property P*, such that if a system S does not exist, then S cannot have property P*?
Assume that P* does exist, S does not exist, and that S has property P*. How would we know that S has property P*, except for the effects of property P* on the observable world? Note that any property imputed by deduction or inference generally must have began with an observation. Therefore, the set of properties that contradict Assumption 1, are the set of measurable properties. In essence, there is only one forbidden property, perhaps abstracted as measurability. In summary, if a system does not exist, then the unique forbidden property of that non-existent system is measurability.
I was looking at pictures of Frank Sinatra’s home in Palm Springs, and I noticed that the kitchen looked very similar to a kitchen that I described in a book I wrote, VeGa. You can just take my word for it since it’s a long book, but the similarities are a bit tough to accept as mere coincidence. Now, it’s entirely possible I previously saw pictures of Sinatra’s home, since I’m a huge fan and have been for my entire adult life, but it doesn’t matter to the conclusion that followed, which is that art in particular and aesthetics generally, create an opportunity for information about a person’s genome to be expressed, and therefore, facilitate mating decisions.
I’ve seen some evidence that preferences are genetic, and this should surprise no one, since people clearly take aesthetic preferences seriously, often motivating people to work harder, just so they can live in a particular home, and wear particular clothes. The more formal conclusion I reached this morning is not on the empirical side, it is instead a theoretical observation, that the expression of preferences creates a language in which information about genomes can be conveyed. There is still of course the empirical question of whether or not this is actually happening, but as we’ll see, the opportunity to convey an enormous amount of information using preferences certainly exists.
Let’s begin by formalizing and limiting preferences as expressed with respect to a particular artifact (e.g., a piece of music, a painting, a pair of pants, etc.), as follows: I hate it, I don’t like it, it’s OK, I like it, and I love it. This creates an ordinal scale of 5 possibilities, ranging from hate to indifference to love, that we’ll use to express preferences. In reality, people have written entire books about single paintings, so the real world range of expression, is much wider, but as we’ll see, it doesn’t matter, you already have an incredibly expressive language using just these five possibilities.
Specifically, for any artifacts, there will be possible rankings. As a consequence, two people discussing, e.g., just 10 works of art, creates 9,765,625 possible rankings, and again, all they have to say is, I hate it, I don’t like it, it’s OK, I like it, or I love it, 10 times, and they’ll convey about 23 binary bits of information, which is genetic bases worth of information. As you can see, increasing the number of possible rankings simply increases the base of the exponent, whereas the exponent dominates the counting function.
Common sense says that people will primarily respond to appearance, smell, taste, and other visceral information when selecting mates, but there are still at least two potentially important roles that art and aesthetics could facilitate in mating decisions, and thereby explain its otherwise anomalous importance to humanity: (1) filtering large crowds of individuals on the basis of shared preferences and (2) marginal distinctions within homogenous populations.
Understanding point (1) is straightforward, if you e.g., put together an event that centers around aesthetic artifacts (e.g., a concert, a gallery exhibition, etc.), then generally speaking, people will attend that event only if they’re sufficiently interested in the full set of artifacts, and the particular combination of artifacts in question. That is, even if you like both Keith Haring and Caravaggio, it’s a bit weird to combine both in a single exhibit.
Therefore, we can express the same ordinal rankings over combinations of artifacts, and that drastically increases the amount of information conveyed. Specifically, given artifacts, there are possible combinations of artifacts (i.e., the cardinality of the set of all subsets of artifacts), each of which is capable of an ordinal ranking, and therefore, we have, for any artifacts, possible rankings over the set of all subsets of those artifacts. If we set as we did above, i.e., considering just 10 artifacts, we find that , which is approximately 1187 genetic bases worth of information, or approximately 7.0% of an entire human mtDNA genome. Now we’re talking about a significant amount of information, that can realistically be conveyed, since , and most people should be able to meaningfully consider around 1000 collections of artifacts, at least over a significant period of time. Just imagine selecting an outfit to wear to a particular gallery exhibition, this is exactly the kind of combination of preferences that we’re considering, which quickly start to cover large numbers of possible combinations. In fact, even having strong preferences of this sort could demonstrate genetic fitness, in addition to conveying genetic information. The net point being, because it’s possible, and it’s obviously something many people take very seriously, it should have some biological function, and I think it’s the obvious: you’re conveying information about your genome through your preferences.
Point (2) is not hard to understand given the above, which is that in a genetically and morphologically homogenous society (i.e., everyone is genetically and physically very similar), marginal differences in genomes could be important, in particular because you have a high risk of incest. It’s not clear to me that there’s any real evidence of heightened selection for aesthetics in homogenous societies, but the discussions above suggest it should happen, because you can convey marginal differences in genetics through your preferences, allowing people to maximize genetic diversity in an otherwise homogenous population.
I was stuck at Neanderthal Genome 8 in my dataset, which you can find here on the NIH website. I simply could not construct a reasonable history for it using the rest of the Neanderthal genomes. However, just like the other presumably misclassified Neanderthal genomes in my dataset, the provenance file (i.e., the previous link) for Neanderthal Genome 8 also contains a qualified entry in that the “isolate” field is set to “Denisova 15”, suggesting again, that this is actually a Denisovan genome, that is somehow associated with Neanderthals. To test this hypothesis, I compared Neanderthal Genome 8 to all other genomes in my dataset to kick the tires from scratch, and I noticed that Neanderthal Genome 8 is a 98.50% match to a modern African genome from Cameroon. That same Cameroon genome, also tests as Denisovan, specifically, it is a 51.09% match to this Denisovan genome. The logical conclusion, is that Neanderthal Genome 8 was similarly misclassified, and is instead yet another Denisovan genome of African origin.
I now have very little doubt about the Out of Africa hypothesis, and specifically, I think the modern day people of Cameroon are related to the first humans, since every test I’ve come up with points to them as the ancestor of all the archaic genomes in the dataset. Since the modern day Cameroon people test as the ancestors of the archaic genomes, they are presumably even more archaic, but somehow still alive. You can read more about this here. I should be done with the complete history of the Neanderthal and Denisovan lines shortly, it’s just a lot of information and much more complicated than Heidelbergensis, which was astonishingly simple and obvious.
I’m building up to a formal paper on human history that uses Machine Learning applied to mtDNA. You can find an informal but fairly rigorous summary that I wrote here [1], that includes the dataset in question and the code. In this note, I’m going to treat the topic at the individual genome level, whereas in [1], I generally applied algorithms to entire populations at a time (i.e., multiple genomes of the same ethnicity), and looked at genetic similarities across entire populations. The goal here is to tell the story of the Heidelbergensis maternal line, which is the largest maternal line in the dataset, accounting for 414 of the 644 genomes in the dataset (i.e., 62.35%). Specifically, 414 genomes are at least a 90% match to either Heidelbergensis itself, or one of the related genomes we’ll discuss below.
The Dataset
The dataset consists of 644 whole mtDNA genomes taken from the NIH database. There are therefore 644 rows, and columns, each column representing a base of the genome stored in that row (i.e., each column entry is one of the bases A, C, G, or T, though there are some missing bases, represented by 0’s). Said otherwise, each genome contains bases, and each row of the dataset contains a full mtDNA genome.
I’ve diligenced the genome provenance files (see, e.g., this Norwegian genome’s provenance file) to ensure the ethnicity of the individual in question is, e.g., a person that is ethnically Norwegian, as opposed to a resident of Norway. The dataset consists of 75 classes of genomes, which are, generally speaking, ethnicities, and column N+1 contains an integer classifier for each genome, representing the ethnicity of the genome (e.g., Norway is represented by the classifier 7). The dataset also contains 19 archaic genomes, that similarly have unique classifiers, that are treated as ethnicities as a practical matter. For example, there are 8 Neanderthal genomes, each of which have a classifier of 32, and are for all statistical tests treated as a single ethnicity, though as I noted previously, Neanderthals are decidedly heterogenous. So big picture, we have 644 full mtDNA genomes, each stored as a row in a matrix (i.e., the dataset), where each of the first columns contains a base of the applicable genome, and an integer classifier in column N+1, that tells you what ethnicity the genome belongs to.
Heidelbergensis and mtDNA
Heidelbergensis is an archaic human that lived (according to Brittanica) approximately 600,000 to 200,000 years ago. When I first started doing research into mtDNA, I immediately noticed that a lot of modern mtDNA genomes were a 95% or more match to Heidelbergensis. I thought at first I was doing something wrong, though I recently proved (both mathematically and empirically) that this is definitely not the case, and in fact, there’s only one way to compare whole mtDNA genomes. You can read the previous note linked to for details, but the short story is, mtDNA is generally inherited directly from your mother (i.e., there’s no paternal DNA at all in mtDNA), with no mutations, though mutations can occur over long periods of time (i.e., thousands of years, or sometimes more).
As a result, any method you use to compare an entire mtDNA genome must be able to produce nearly perfect matches, since a large enough dataset should contain a basically perfect match for a significant number of genomes, given mtDNA’s extremely slow rate of mutation. Said otherwise, if you have a large number of whole mtDNA genomes, there should be nearly perfect matches for a lot of the genomes in the dataset, since mtDNA mutates extremely slowly. There are of course exceptions, especially when you’re working with archaic genomes that might not have survived to the present, but the gist is, mtDNA mutates so slowly, someone should have basically the same mtDNA as you. Empirically, there’s exactly one method of whole-genome comparison that accomplishes this, which is explained in the previous link and contains the applicable code to test the hypothesis.
Just in case it’s not clear, whole-genome comparison means you take two entire genomes, and compare them side-by-side, rather than looking for individual sequential segments like genes, which until recently, was the more popular approach. If you’re curious, I’ve demonstrated that whole-genome comparison, and random base selection, are categorically superior to relying on sequential bases (e.g., genes) for imputation, at least as applied to mtDNA. See, A New Model of Computational Genomics [2]. We will also discuss using genome segments in the final section below.
A Heidelbergensis skull, image courtesy of Britannica.
Whole-Genome Comparison
The method of comparison that follows from this observation is straight forward, you simply count the number of matching bases between two genomes. So for example, if we’re given genome and , the number of matching bases is simply 2. Because mtDNA is circular, it’s not clear where to start the comparison. For example, we could start reading genome at the first , rather than the first base . However, the previous link demonstrates that there’s exactly one whole-genomealignment (otherwise known as a global alignment), or starting index for mtDNA, the rest of them are simply not credible for the reasons discussed above.
This makes whole-genome comparison super easy, and incredibly fast, and in fact, my software can compare a given genome to all 644 genomes in the dataset in just 0.02 seconds, running on an Apple M2 Pro, producing a ton of statistics for the input genome, not just the number of matching bases. Sure, it’s a great machine, but it’s not a super computer, which means now everyone can do real genetic analysis on consumer devices. Once popularized, these methods will probably make short work of the complete history of mankind, and possibly the entire history of life itself, since mtDNA is not unique to humans. Further, these methods and their results are rock solid, empirical evidence for the Theory of Evolution, which as you’ll see below, is not subject to serious criticism, at least with respect to mtDNA.
Modern Relatives of Heidelbergensis
As noted above, many modern living humans have mtDNA that is a 95% or more match to the single Heidelbergensis genome in the dataset. The genome was found at Sima de Los Huesos, and there is apparently some debate about whether it is actually a Neanderthal, but it is in all cases a very archaic genome from around 500,000 years ago. As such, though I concede this Heidelbergensis genome is a 95.10% match to the third Neanderthal genome in my dataset, which is from around 100,000 years ago, I think it’s best to distinguish between the two, given the huge amount of time between the two genomes, and the fact that they’re not exactly the same genome.
Recall that we are comparing whole-genomes by simply counting the number of matching bases, which we’ll call the match count. We can therefore, set a minimum match count of say , i.e., 90% of the genome, and retrieve all genomes that are at least a 90% match to Heidelbergensis. This produces the chart below, where the height of the bar provides the percentage of genomes in the applicable population that are at least a 90% match to the single Heidelbergensis genome. For example, 100% of the Iberian Romani are at least a 90% match to the Heidelbergensis genome, producing a height of 1.0 in the chart below. The population acronyms can be found at the back of [2], but just to highlight some of the obvious matches, KZ stands for Kazakhstan, IB stands for Iberian Romani, IT stands for Italy, and RU stands for Russia.
A chart showing the percentage of each population that is at least a 90% match to Heidelbergensis.
The plain takeaway is that many modern humans carry mtDNA that is close to Heidelbergensis, peaking at a 96.69% match for a Kazakh individual. As noted above, when working with modern genomes, you’ll often find a basically perfect match that exceeds 99%, but when working with archaic genomes, that’s not always the case, and it makes perfect sense, since so much time has elapsed, that even with the incredibly slow rate of mutation for mtDNA, a few percentage points of mutation drift is to be expected.
The Phoenician People
The Phoenicians were a Mediterranean people that existed from around 2500 BC to 64 AD. Though there could be other example genomes, the Phoenicians are a great case study because they are a partial match to Heidelbergensis, and a partial match to the Pre-Roman Ancient Egyptian genome. You can already appreciate the intuition, that Heidelbergensis evolved into the Phoenicians, and then the Phoenicians evolved further into the Ancient Egyptians.
Now the real story is more complicated, and it doesn’t look like all of this happened in the Mediterranean. Instead, it looks like human life begins in West Africa, migrates to roughly the Mediterranean and Eurasia, migrates further to somewhere around Northern India, and then spreads back to Europe and Africa, and further out into East Asia. You can read [2] for more on this topic, this note will instead be focused on the evolution of the individual genomes, and less so on claims regarding their historical geographies. That is, I’m going to present you with a set of genomes that begin with Heidelbergensis, and end in the Icelandic people, who are almost certainly Vikings, but I’m not going to argue too much about where these mutations happened, outside of a few notes for context, so that it’s not all happening in a void.
Returning to the Phoenicians, we want to show first, that the Phoenicians evolved from Heidelbergensis. All of these steps will involve epistemological reflections, so that we can be comfortable that we’re asserting reasonable claims. That said, as you’ll see, all of these claims are uncertain, and plainly subject to falsification, but that’s science. To begin, note that there are 6 Phoenician genomes in the dataset, and that the first Phoenician genome in the dataset (row 415) is at least a 99.72% match to the other 5 Phoenician genomes. As such, to keep things simple, we will treat this first Phoenician genome as a representative genome for the entire class of Phoenicians. Further, note that the first Phoenician genome is a 41.17% match to Heidelbergensis. If we were comparing two random genomes, then the expected match count is 25% of the genome, since the distribution is given by the Binomial Distribution, with a probability of success of . That is, at each base, we have two random variables, one for each genome, and each of those variables can take on a value of A, C, G, or T. If it’s truly random, then there are possible outcomes, and only 4 of those outcomes correspond to the bases being the same, producing a probability of . Therefore, we can conclude that the match count of 41.17% between Heidelbergensis and the Phoenician genome is probably not the result of chance.
The claim that the two genomes are truly related finds further support in the location of the matching bases, which are concentrated in the first 3,500 bases, which is shown in the chart below. The chart below is produced by taking 500 bases at a time, starting with the first 500 bases of each genome, and counting how many bases within that 500-base segment match between the two genomes. The maximum match count is of course 500 bases, which would produce a height of 1.0, or 100%. This process continues over the entire genomes, producing the chart below. As you can see, the most significant matches are clustered in the first 7 segments, representing the first 3,500 bases of the genomes. The argument is, because there is a significant, contiguous segment within the genomes that are highly similar, we can confidently rule out chance as the driver of the similarity. You can never be totally certain, but since it’s probably not chance that’s driving the similarity, the logical conclusion is that heredity and mutation is what caused the similarity between the two genomes. Now we don’t know the direction of time from this analysis alone (i.e., either genome could have evolved into the other), but because Heidelbergensis is very archaic, the logical conclusion, is that Heidelbergensis mutated, eventually forming the Phoenician maternal line.
A chart showing the percentage of matching bases between the Heidelbergensis and Phoenician genome, broken into 500-base segments.
One important point to note, is that even if a genome evolves, it does not imply that all instances of that genome evolve. For example, as noted above, 100% of the living Iberian Romani people are at least a 90% match to Heidelbergensis, demonstrating that at least some Heidelbergensis genomes did not evolve into the Phoenician line, and instead remained roughly the same over time. As such, we can say confidently that mtDNA is very slow to mutate as a general matter, but the rates of mutation are heterogenous.
Just to close this section with some context for modern humans that carry the Phoenician line, 80% of living Sardinians and 33.33% of living Vedda Aboriginals are at least a 90% match to the Phoenicians. Obviously, it’s a bit shocking that you’d have Phoenician mtDNA in Asia, but if you read [2], you’ll quickly learn that these are global maternal lines that often contain multiple disparate people. Two common sense explanations, (1) the Phoenicians really made it to Asia or (2) there’s a common ancestor for both the Phoenician and Vedda people, presumably somewhere in Asia. Hypothesis (2) finds support in the fact that 10.52% of Mongolians are also at least a 90% match to the Phoenicians. This is a complicated topic, and it’s just for context, the real point of this note is that you can plainly see that Heidelbergensis evolved, which is already interesting and compelling evidence for the Theory of Evolution, and specifically, it evolved into the Phoenician maternal line.
The Ancient Egyptians
Introduction
The Ancient Egyptians were a Mediterranean civilization that lasted from around 3150 BC to 30 BC, until it was ruled by Rome, from around 30 BC to 642 AD. There are two Ancient Egyptian genomes in the dataset, one from approximately 2000 BC, before Roman rule, and another genome from approximately 129 to 385 AD, during Roman rule. This is a huge amount of time, and so it’s not surprising that the demographics changed. But the Ancient Egyptians present a shocking demographic shift, from earlier rulers that were plainly of Asian origin, to rulers that looked, and were known to be, European. For example, see the panel of images below, with Nefertiti (1353 to 1336 BC) on the left, then King Menkaure and his Queen (2550 BC to 2503 BC), and finally Cleopatra (51 to 30 BC) on the right, who is known to be Macedonian.
Queen Nefertiti, image courtesy of Wikipedia.King Menkaure and Queen, image courtesy of Museum of Fine Arts Boston.Queen Cleopatra, image courtesy of Wikipedia.
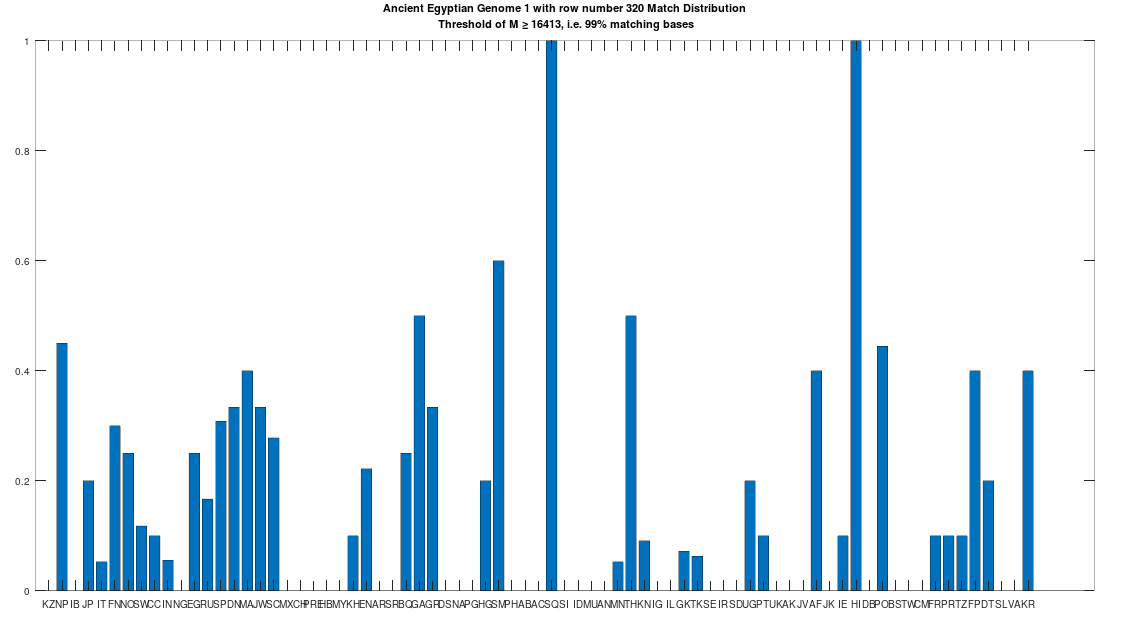
The hypothesis that the earlier Egyptians were of Asian origin is further supported by the chart below, which shows the distribution of genomes that are at least a 99% match to the Pre-Roman Egyptian genome. The full set of population acronyms are in [2], but for now note that NP stands for Nepal, JP stands for Japan, FN stands for Finland, NO stands for Norway, EG stands for modern day Egypt, DN stands for Denmark, GA stands for Georgia, TH stands for Thailand, FP stands for Philippines, and KR stands for Korea. As you can plainly see, the Pre-Roman Egyptian genome is very common in Northern Europe and East Asia, with very little representation in Africa outside of modern day Egypt, though there is some nuance to this. See [2] for more. The point is, the Pre-Roman Egyptian genome probably comes from Asia, and spread to Northern Europe, North Africa, and East Asia, and as far as I know, this is not exactly accepted history, but it’s clearly the case.
A chart showing the percentage of each population that is at least a 99% match to the Pre-Roman Egyptian genome.
Ancestry from Heidelbergensis and Phoenicia
As noted above, the Pre-Roman Egyptian genome (row 320) is a partial match to the Phoenician genome, with a match count of 88% of the genome. This is obviously very high, so we can be confident that this is not the result of chance, and is instead the result of heredity and mutation. Further, because we have assumed that Heidelbergensis is the ancestor of the Phoenician genome (since it is archaic), it cannot be the case that the Ancient Egyptian genome is also the ancestor of the Phoenician genome. Specifically, because mtDNA is inherited directly from the mother to its offspring, there can be only one ancestral maternal line for a given genome, though there can be intermediate ancestors. For example, genome A mutates into genome B, which in turn mutates into genome C. However, because the Ancient Egyptian genome has a match count of 29.73% to Heidelbergensis, the Ancient Egyptian genome cannot credibly be an intermediate ancestor of the Phoenicians, between Heidelbergensis and the Ancient Egyptians. Therefore, it must be the case, given our assumption that Heidelbergensis is the ancestor of the Phoenicians, that the Pre-Roman Ancient Egyptian genome is the descendant of the Phoenicians.
Historically, this is counterintuitive, because the Ancient Egyptians are more ancient than the Phoenicians, but as noted above, these maternal lines are broader groups, I’m simply labelling them by using the most famous civilizations that have the genomes in question. Further, as noted above, a lot of this evolution probably happened in Asia, not the Mediterranean. So one sensible hypothesis is that Heidelbergensis travelled East, mutated to the Phoenician line somewhere in Asia, and then that Phoenician line mutated further into the Pre-Roman Ancient Egyptian line, again probably somewhere in Asia. This is consistent with the fact that 76.67% of Kazakh genomes, 44.44% of Indian genomes, and 66.67% of Russian genomes are at least a 95% match to Heidelbergensis, making it plain that Heidelbergensis travelled to Eurasia and Asia. In contrast, as noted above, the Pre-Roman Ancient Egyptian line is found generally in Northern Europe, East Asia, and North Africa, consistent with a further migration from Eurasia and Asia, into those regions.
The Roman Era Egyptian Genome
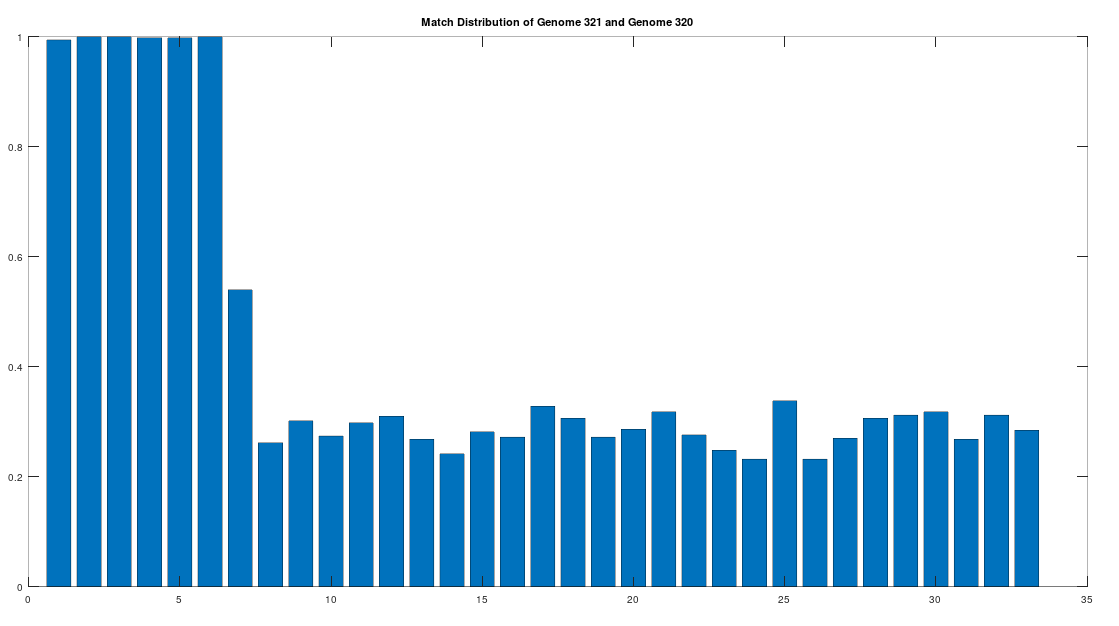
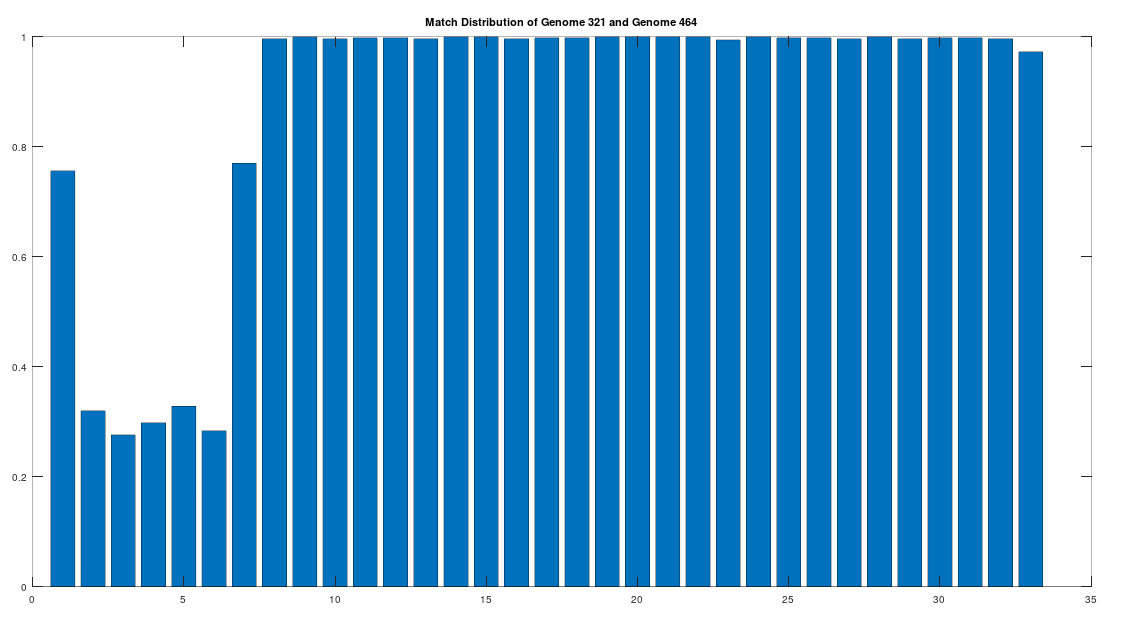
As noted above, the Ancient Egyptians were ruled by Rome from around 30 BC to 642 AD. Though it is reasonable to assume that there were resultant demographic changes, we’re only looking at two genomes from Ancient Egypt, and so the point is not that these two genomes are evidence of that demographic change. The evidence of the demographic changes are above, in the form of archeological evidence of completely different people ruling their civilization. The point of this section is instead that there is a second genome that was found in Egypt, that is dated to around 129 to 385 AD, squarely during Rome’s rule over Egypt, that is related to the other Ancient Egyptian genome discussed above. Specifically, the Roman Era Egyptian genome (row 321) is a 42.20% match to the Pre-Roman Egyptian genome (row 320). Now, that is significantly above chance (i.e., 25%), but we can also perform the same analysis we did above, looking to 500-base segments for confirmation that the match count is not the result of chance, which is shown below. Again, the most similar regions are concentrated in the first seven, 500-base segments, plainly suggesting heredity rather than chance.
Because we have assumed the Phoenician genome is the ancestor of the Pre-Roman Egyptian genome, it cannot be the case that the Roman Era Egyptian genome is the ancestor of the Pre-Roman Egyptian genome. We can further rule out the possibility of an intermediate relationship by noting that the match count between the Roman Era Egyptian genome and the Phoenician genome is 30.50%. Therefore, we have established a credible claim that Heidelbergensis evolved into the Phoenician maternal line, which in turn evolved into the Pre-Roman Egyptian maternal line, and then further into the Roman Era Egyptian maternal line.
Iceland and the Vikings
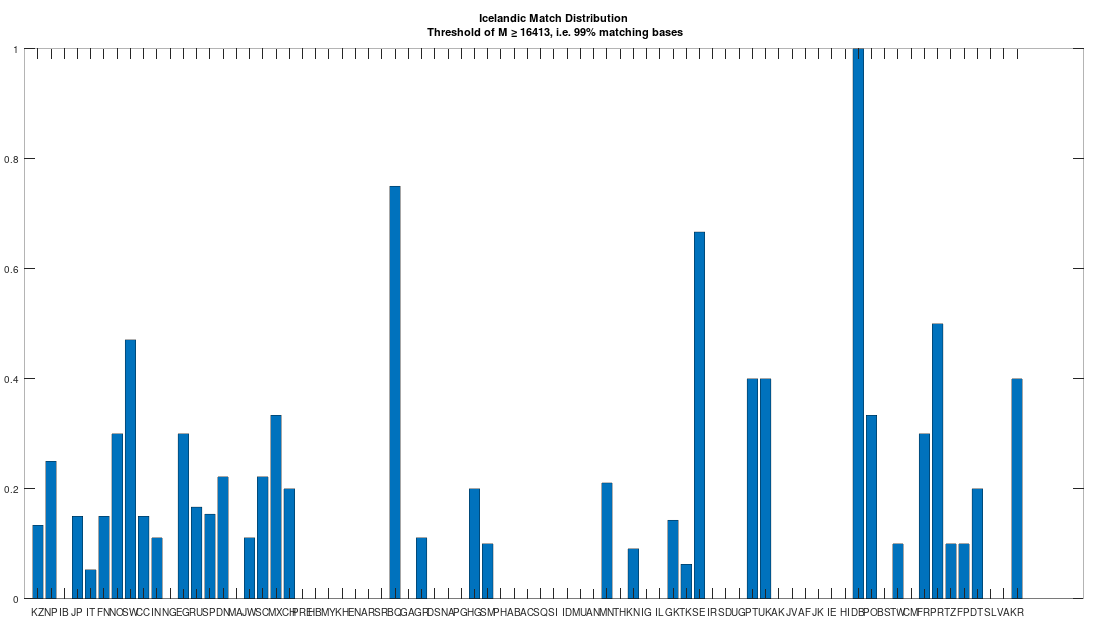
The dataset contains a single Icelandic genome, though it was collected from a person in Canada. So it’s fair to express some skepticism, as people can deliberately deceive researchers, though I’m not sure why you would. But that said, let’s kick the tires, and see what populations are at least a 99% match to this purportedly Icelandic genome, which is shown in the chart below. Because these genomes are members of large global groups, we need to be careful in this type of analysis, and accept uncertainty. But you can plainly see in the chart below, that the genome in question is a pronounced match to Sweden (SW) and Norway (NO). Further, the Icelandic genome is a 99.77% match to the single Dublin genome (DB), and Dublin was a Viking colony. Now all of this is subject to falsification and uncertainty, but I think we can be reasonably confident, that the person in question really is of Icelandic ancestry.
A chart showing the percentage of each population that is at least a 99% match to the Icelandic genome.
With that, we can turn to heredity, in particular noting that the Roman Era Egyptian genome is an 87.79% match to the Icelandic genome. Though that is an extremely high match count, that cannot credibly be the result of chance, we can also examine the structure of the matching segments, just as we did above, since we have some doubt regarding the provenance, given that the individual lived in Canada. This is shown below, and as you can see, it is plainly not the result of chance, since the vast majority of matching segments are from and including segment 7 onward. Because we have assumed that the Pre-Roman Egyptian genome is the ancestor of the Roman Era Egyptian genome, it cannot be the case that the Icelandic genome is the ancestor of the Roman Era Egyptian genome. To rule out an intermediate relationship, we can simply note that the match count between the Icelandic genome and the Pre-Roman Egyptian genome is 30.29%. Therefore, we have put together a credible claim that Heidelbergensis evolved into the Phoenician maternal line, which in turn produced the Pre-Roman Egyptian maternal line, then the Roman Era Egyptian maternal line, and finally, the Icelandic maternal line. Because Iceland was uninhabited before the Vikings, it is reasonable to assume that the Icelandic genome was included in the set of Viking maternal lines.
A chart showing the percentage of matching bases between the Roman Era Egyptian genome and the Icelandic genome, broken into 500-base segments.
Measuring Genetic Drift
As you can see, whole-genome comparison is nothing short of amazing, allowing us to build rock solid arguments regarding the history of mankind, and demonstrating plainly the Theory of Evolution is real. That said, if a genome is subject to what’s called an indel, which is an insertion or deletion, then the match count between two genomes will generally drop to around 25%, i.e., chance. As a simple example, consider genomes and . These two genomes have a match count of 3 bases, or 75% of the genome. Now let’s say we create an indel in genome , inserting a “G” after the first “A”, producing the genome . The match count is now instead 1 base, or 25% of the genome, depending on which genome’s length you use (i.e., 4 or 5).
As a result, geneticists make use of what are called local alignments, which take segments from one genome, and find the best match for that segment in the comparison genome. Continuing with and , a local alignment could, e.g., take the segment from , and map it to in genome , producing a match count of 2 bases. The algorithm I’ve put together does exactly this, except using 500-base segments from an input genome, searching for the best match for that segment in the comparison genome. During this process, the algorithm also identifies, and counts the number of insertions and deletions that have occurred between the two genomes (i.e., the total number of indels). The indel count provides us with a second measure of genetic drift, in addition to the match count, which is still produced by the local alignment algorithm, and is given by the total number of matches across all 500-base segments. That is, the match count for a local alignment, is the sum of all the match counts for the segments, where each segment has a maximum match count of 500 bases.
Applying this to the narrative above, we can run the local alignment algorithm comparing Heidelbergensis to the Icelandic genome. This produces a match count of 15,908, and therefore, mutations occurred over the entire history outlined above, which is not that many, since it spans around 500,000 years. Further, the local alignment algorithm found only 2 indels between the two genomes. This is all consistent with the extremely slow rate of mutation of mtDNA generally. That said, note that unlike the whole-genome algorithm, the local alignment algorithm is approximate, since there is (to my knowledge) no single segment length (in this case 500 bases) that is an objective invariant for comparing two genomes. Said otherwise, when using whole-genome comparison, both mathematical theory and empiricism show there’s only one global alignment, and therefore only one algorithm that gets the job done. In contrast, local alignments can produce different results if we vary the segment length, which is again in this case set to 500 bases. But the bottom line is, there really isn’t that much change over a huge period of time.
Dark Matter is predicted to exist because cosmological observations deviate from Relativity. Now, real mathematicians and scientists have garbage cans, and when observation deviates from predictions produced by a model, we throw the model in the garbage. So the best explanation for Dark Matter, is that it doesn’t exist, and Relativity is wrong. We already know that Relativity is wrong for other reasons, e.g., the existence of the Neutrino, which has mass, yet a velocity of c, which is completely impossible in Relativity. But rather than accept this, ostensible scientists fuss about whether it’s exactly c, or some other handwaving nonsense, in defense of a physically implausible theory of the Universe, where everything depends upon anthropomorphic observers, as if electrons had a brain. So I think the best answer for Dark Matter, is that Relativity is wrong, and we need a new model of physics to explain cosmological observations.
In contrast, I don’t think Dark Energy depends upon Relativity. If however, it does depend upon Relativity, then we should start with the assumption that Dark Energy doesn’t exist, and build a new model of physics that is consistent with cosmological observations, otherwise known as doing science. All of that said, it dawned on me last night, that there might be something to Dark Energy, specifically, if we complete the symmetry of gravity.
Let’s start by completing the symmetry of gravity, which requires positive and negative mass, which we will denote as and , respectively. Let’s further assume that we are surrounded almost exclusively by positive mass, which again is denoted by . Let’s assume that positive mass is attracted to positive mass, which would obviously be perfectly consistent with gravity, since we’ve already assumed that almost all matter we are exposed to is positive mass. Further, let’s assume that negative mass is similarly attracted to negative mass, and that masses with opposite signs cause repulsion. That is, given some quantity of positive mass and negative mass , the two masses will repel each other when sufficiently proximate. This completes the symmetry of gravity, since we now have positive and negative masses, and attractive and repulsive forces.
The obvious question is, where’s all this negative mass, and repulsive force? Well, first let’s consider that the Universe is quite old, and as a result, if we posit such a force has always existed, then this would eventually divide the Universe into at least two regions, one filled with mostly positive mass, and one filled with mostly negative mass. If this process is less than complete, then we should see some inexplicable acceleration due to interactions between positive and negative masses, which is consistent with Dark Energy.
Now, the SLAC 144 Experiment demonstrates that light on light collisions can produce matter-antimatter pairs. Further, we know that matter-antimatter collisions can create light. Let’s assume again that we’re in a positive matter region of the Universe, and as such, if we allow to represent a photon, we could write that , to represent a positron and electron colliding to form a photon. Similarly, we could write that to represent a light on light collision producing an electron-positron pair.
With that notation, now let’s posit the negative-mass versions of the electron and positron, which we will denote as and . The question is, what do we get when we combine the negative-mass versions of an electron and positron? Expressed symbolically, , where is some unknown particle.
If it’s an ordinary photon, then we arguably have a problem, because photon collisions have been shown to produce positive-mass electron positron pairs. That is, under this hypothetical, in the negative-mass regions of the Universe, when you do light on light collisions, you still get positive-mass pairs, which should eventually be kicked out of that region of space. In contrast, in the positive-mass regions of the Universe, when you do light on light collisions, you also get positive-mass pairs, but they stick around because it’s ordinary mass.
We could hypothesize that this is just the way things are, but we get an elegant potential solution to the source of Dark Energy if we instead posit the existence of negative-mass light , as a particle distinct from ordinary light. Specifically, if we further posit that negative-mass light does not interact with positive mass, and that positive-mass light (our ordinary photons) do not interact with negative mass, then that negative mass would not be detectable using ordinary photons. Ironically, this is a kind of “dark” matter, but it’s not the nonsense hypothesized hitherto.
My work on mtDNA has led to a thesis that human life begins in Africa, spreads to Asia, and then spreads (1) back West to Europe and Africa and (2) further East into East Asia and the Pacific. I call this the Migration-Back Hypothesis, and you can read all about it here [1], and here, and on my blog generally, where you’ll find a ton of material on topic.
One of the most interesting observations in my work is that the living modern day people of Cameroon test as having the most ancient genomes in the dataset of complete human mtDNA genomes I’ve assembled, which contains 19 archaic mtDNA genomes, that are Heidelbergensis (1 genome), Neanderthal (10 genomes), and Denisovan (8 genomes). This is not too shocking, considering that 53.01% of the 664 genomes in the dataset are at least a 60% match, to at least one archaic genome. This comparison to the archaic genomes is done using the only sensible global alignment for mtDNA, so you can’t argue that it’s chance, or cherry picking, there are a lot of living people that have archaic mtDNA. The reason I’m writing this note is because I think two of the Neanderthal genomes were misclassified by the scientists that sequenced the genomes.
I’ve written previously that the Neanderthals are decidedly heterogenous on the maternal line, in that there are 10 Neanderthal genomes, that can be broken into 6 completely distinct clusters (i.e., groups of similar genomes). I’m using a global alignment for all of this work, except where noted below, and as noted above, there’s only 1 sensible global alignment for mtDNA, so these distinctions are objective.
Specifically, (i) genomes 1, 2, and 10 are at least a 99.5% mutual match to each other, (ii) genomes 5 and 6 are a 63.4% match to each other, (iii) genomes 8 and 9 are a 99.9% match to each other, and (iv) genomes 3, 4, and 7 are unique, and have no meaningful match to each other or the rest of the Neanderthal genomes. This note focuses on genomes 5 and 6, which appear to be misclassified as Neanderthals, and instead seem to be Denisovans based upon their mtDNA. All of the provenance files for the relevant genomes are linked to below at the bottom of the article, and each provenance file includes a FASTA file that contains the applicable full genome. The full dataset I’ve assembled (which includes all of these archaic genomes) is available in [1] above.
Neanderthal Genome 5
The provenance file for Neanderthal Genome 5 (row 389 of my dataset) lists the “organism” field as “Homo sapiens neanderthalensis”, and the “sub_species” field as “neanderthalensis”. However, the genome title includes the phrase “Denisova 17″, and the “isolate” field is listed as “Denisovan 17”. Further, the article associated with the genome suggests that the genome is actually from the Denisova Cave in Siberia, yet they classified it as Neanderthal, which doesn’t look right. The relevant quote is on page 30 (page 3 of the pdf):
We estimated the molecular age of the mtDNA of the newly identified Neanderthal (Denisova 17) to ~134 ka (95% height posterior density (HPD): 94–177 ka) using Bayesian dating…
Note that “Denisovan 17” is a label used by the authors of the quoted article, I’m using indexes and row numbers keyed to my dataset (i.e., “Denisovan 17” is Neanderthal Genome 5 in my dataset). However, as noted above, Neanderthal Genome 5 is a 63.4% match to Neanderthal Genome 6 only, and is not a significant match to any other Neanderthal genome. This suggests that these two genomes are, as noted above, a distinct maternal line that lived among other maternal lines, that have all been archeologically classified as Neanderthals. However, Neanderthal Genome 5 was found in the Denisovan Cave in Siberia, per the article quoted above, which is already evidence for the claim that it is actually a Denisovan, at least with respect to its maternal line.
Further, Neanderthal Genome 5 has 8,915 bases (i.e., 53.77% of the full genome) in common with Denisovan Genome 1 (row 377 of my dataset), using the whole genome global alignment, which is well beyond chance (i.e., 25.00% of the full genome). In contrast, Neanderthal Genome 5 has 5,300 bases (i.e., 31.96% of the full genome) in common with its closest match among the other Neanderthal Genomes (save for Neanderthal Genome 6, which also seems to be Denisovan, and is discussed below).
Finally, Neanderthal Genome 5 has 16,328 bases (i.e., 98.48% of the full genome) in common with a Cameroon Genome (row 591 of my dataset). That Cameroon Genome in turn has 8,898 bases (i.e., 53.47% of the full genome) in common with the same Denisovan Genome 1 (row 377 of my dataset). The plain conclusion is that Neanderthal Genome 5 is an archaic Siberian Denisovan individual, with a close maternal connection to living West Africans. As noted above, the Cameroon test as the most ancient people across my dataset, suggesting a migration from Cameroon to Siberia, which is consistent with the Out of Africa Hypothesis, but does not contradict my Migration-Back Hypothesis, since it’s entirely possible that later Denisovans migrated back to Europe or Africa from Siberia, or further into East Asia and the Pacific. However, that is not the point of this note, which is limited to the misclassification of two Neanderthal genomes.
Neanderthal Genome 6
Similarly, Neanderthal Genome 6 has 5,289 bases (i.e., 31.90% of the full genome) in common with its closest match among the other Neanderthal Genomes (save for Neanderthal Genome 6, which also seems to be Denisovan, as discussed above). In contrast, Neanderthal Genome 6 has 8,588 bases (i.e., 51.80% of the full genome) in common with Denisovan Genome 1 (row 377 of my dataset). Further, Neanderthal Genome 6 has 10,461 bases (i.e., 63.09% of the full genome) in common with the same Cameroon genome discussed above. However, unlike Neanderthal Genome 5, the provenance file for Neanderthal Genome 6, and the related article, make it clear the genome was discovered in Scladina, which is an archeological site in Belgium. Even using a local alignment, the resultant number of matching bases between Neanderthal Genome 6 and the Cameroon genome is 16,183, which is lower than the number of matching bases between Neanderthal Genome 6 and that same genome (i.e., 16,328) using a global alignment. Note that local alignments maximize the number of matching bases. The sensible conclusion being that Neanderthal Genome 6 is actually Denisovan, though it is not as close to the Cameroon genome as Neanderthal Genome 5, though it is close enough to infer African ancestry. This is again consistent with the Out of Africa Hypothesis, though it’s not clear whether this genome has any connection to Asia, at least limited to this discussion alone, and as such, it adds no further credibility to my Migration-Back Hypothesis, though it does not contradict the Migration-Back Hypothesis in any way, since it’s entirely possible at least some people left Africa directly for Europe or other places. In contrast, the Migration-Back Hypothesis is about the overall migration patterns of some of the most modern mtDNA genomes in the dataset, linking otherwise disparate modern humans across enormous distances.
Because of relatively recent advances in genetic sequencing, we can now read entire mtDNA genomes. However, because mtDNA is circular, it’s not clear where you should start reading the genome. As a consequence, when comparing two genomes, you have no common starting point, and the selection of that starting point will impact the number of matching bases. As a simple example, consider the two fictitious genomes and . If we count matching bases using the first index of each genome, then the number of matching bases is zero. If instead we start at the first index of and the second index of (and loop back around to the first ‘G’ of ), the match count will be four, or 100% of the bases. As such, determining the starting indexes for comparison (i.e., the genome alignment) is determinative of the match count.
It turns out that mtDNA is unique in that it is inherited directly from the mother, generally without any mutations at all. As such, the intuition for combinations of sequences typically associated with genetics is inapplicable to mtDNA, since there is no combination of traits or sequences inherited from the mother and the father, and instead a basically perfect copy of the mother’s genome is inherited. As a result, it makes perfect sense to use a global alignment, which we did above, where we compared one entire genome to another entire genome . In contrast, we could instead make use of a local alignment, where we compare segments of two genomes.
For example, consider genomes and . First you’ll note these genomes are not the same length, unlike in the example above, which is another factor to be considered when developing an alignment for comparison. If we simply use the first three bases of each genome for comparison, then the match count will be one, since the first two initial ‘A’s match. If instead we use index two of and index one of , then the entire sequence matches, and the resultant match count will be three.
Note that the number of possible global alignments is simply the length of the genome. That is, when using a global alignment, you “fix” one genome, and “rotate” the other, one base at a time, and that will cover all possible global alignments between the two genomes. In contrast, the number of local alignments is much larger, since you have to consider all local alignments of each possible length. As a result, it is much easier to consider all possible global alignments between two genomes, than local alignments. In fact, it turns out there is exactly one plausible global alignment for mtDNA, making global alignments extremely attractive in terms of efficiency. Specifically, it takes 0.02 seconds to compare a given genome to my entire dataset of roughly 650 genomes using a global alignment. Performing the same task using a local alignment takes one hour, and the algorithm I’ve been using considers only a small subset of all possible local alignments. That said, local alignments allow you to take a closer look at two genomes, and find common segments, which could indicate a common evolutionary history. This note discusses global alignments, I’ll write something soon that discusses local alignments, as a second look to support my work on mtDNA generally.
Nearest Neighbor
The Nearest Neighbor algorithm can provably generate perfect accuracy for certain Euclidean datasets. That said, DNA is obviously not Euclidean, and as such, the results I proved do not hold for DNA datasets. However, common sense suggests we might as well try it, and it turns out, you get really good results that are significantly better than chance. To apply the Nearest Neighbor algorithm to an mtDNA genome , we simply find the genome that has the most bases in common with , i.e., its best match in the dataset, and hence, its “Nearest Neighbor”. Symbolically, you could write . As for accuracy, using Nearest Neighbor to predict the ethnicity of each individual in my dataset produces an accuracy of 30.87%, and because there are 75 global ethnicities, chance implies an accuracy of . As such, we can conclude that the Nearest Neighbor algorithm is not producing random results, and more generally, produces results that provide meaningful information about the ethnicities of individuals based solely upon their mtDNA, which is remarkable, since ethnicity is a complex trait, that clearly should depend upon paternal ancestry as well.
The Global Distribution of mtDNA
It turns out the distribution of mtDNA is truly global, and a result, we should not be surprised that the accuracy of the Nearest Neighbor method as applied to my dataset is a little low, though as noted, it is significantly higher than chance and therefore plainly not producing random predictions. That is, if we ask what is e.g., the best match for a Norwegian genome, you could find that it is a Mexican genome, which is in fact the case for this Norwegian genome. Now you might say this is just a Mexican person that lives in Norway, but I’ve of course thought of this, and each genome has been diligenced to ensure that the stated ethnicity of the person is e.g., Norwegian.
Now keep in mind that this is literally the closest match for this Norwegian genome, and it’s somehow on the other side of the world. But high school history teaches us about migration over the Bering Strait, and this could literally be an instance of that, but it doesn’t have to be. The bottom line is, mtDNA mutates so slowly, that outcomes like this are not uncommon. In fact, by definition, because the accuracy of the Nearest Neighbor method is 38.07% when applied to predicting ethnicity, it must be the case that 100% – 38.07% = 69.13% of genomes have a Nearest Neighbor that is of a different ethnicity.
One interpretation is that, oh well, the Nearest Neighbor method isn’t very good at predicting ethnicity, but this is simply incorrect, because the resultant match counts are almost always over 99% of the entire genome. Specifically, 605 of the 664 genomes in the dataset (i.e., 91.11%) map to a Nearest Neighbor that is 99% or more identical to the genome in question. Further, 208 of the 664 genomes in the dataset (i.e., 31.33%) map to a Nearest Neighbor that is 99.9% or more identical to the genome in question. The plain conclusion is that more often than not, nearly identical genomes are found in different ethnicities, and in some cases, the distances are enormous.
In particular, the Pashtuns are the Nearest Neighbors of a significant number of global genomes. Below is a chart showing the number of times (by ethnicity) that a Pashtun genome was a Nearest Neighbor of that ethnicity. So e.g., returning to Norway (column 7), there are 3 Norwegian genomes that have a Pashtun Nearest Neighbor, and so column 7 has a height of 3. More generally, the chart is produced by running the Nearest Neighbor algorithm on every genome in the dataset, and if a given genome maps to a Pashtun genome, we increment the applicable column for the genome’s ethnicity (e.g., Norway, column 7). There are 20 Norwegian genomes, so of Norwegian genomes map to Pashtuns, who are generally located in Central Asia, in particular Afghanistan. This seems far, but in the full context of human history, it’s really not, especially given known migrations, which covered nearly the whole planet.
The chart above is not normalized to show percentages, and instead shows the integer number of Pashtun Nearest Neighbors for each column. However, it turns out that a significant percentage of genomes in ethnicities all over the world map to the Pashtuns, which is just not true generally of other ethnicities. That is, it seems the Pashtuns are a source population (or closely related to that source population) of a significant number of people globally. This is shown in the chart below, which is normalized by dividing each column by the number of genomes in that column’s population, producing a percentage.
As you can see, a significant percentage of Europeans (e.g., Finland, Norway, and Sweden, columns 6, 7, and 8 respectively), East Asians (e.g., Japan and Mongolia, columns 4 and 44, respectively), and Africans (e.g., Kenya and Tanzania, columns 46 and 70, respectively), have genomes that are closest to Pashtuns. Further, the average match count to a Pashtun genome over this chart is , so these are plainly meaningful, nearly identical matches. Finally, these Pashtun genomes that are turning up as Nearest Neighbors are heterogeneous. That is, it’s not the case that a single Pashtun genome is popping up globally, and instead, multiple distinct Pashtun genomes are popping up globally as Nearest Neighbors. One not-so-plausible explanation that I think should be addressed is the Greco-Bactrian Kingdom, which overlaps quite a bit with the geography of the Pashtuns. The hypothesis would be that Ancient Greeks brought European mtDNA to the Pashtuns. Maybe, but I don’t think Alexander the Great made it to Japan, so we need a different hypothesis to explain the global distribution of Pashtun mtDNA.
All of this is instead consistent with what I’ve called the Migration-Back Hypothesis, which is that humanity begins in Africa, migrates to Asia, and then migrates back to Africa and Europe, and further into East Asia. This is a more general hypothesis that many populations, including the Pashtuns, migrated back from Asia to Africa and Europe, and extended their presence into East Asia. The question is, can we also establish that humanity began in Africa using these and other similar methods? Astonishingly, the answer is yes, and this is discussed at some length in a summary on mtDNA that I’ve written.
The vast majority of my work in mtDNA has focused on global alignments, because of the unbelievable efficiency this creates, even when working on consumer devices. However, I’ve basically exhausted the topic of global alignments, so I’ve started to focus on local alignments to further support my research. That is, I’m going to second guess my work, using local alignments, to see if I get the same results. So far, that is exactly the case, using the attached local alignment algorithm, which is very straight forward.
Specifically, it takes a given input genome, and compares it to a comparison genome, by taking 500 bases at a time, and searching one by one for the index of the comparison genome where the match count between those 500 bases from the input genome is maximized, when compared to the comparison genome. It does so for all 500 base segments of the input genome, producing a starting index for each such 500 base segment of the input genome, that is mapped to the comparison genome (i.e., an alignment), and a total match count using that alignment.
It is incomparably slower than my global alignment algorithm, which takes just 0.02 seconds to the find the nearest neighbor of an input genome over a dataset of approximately 650 whole mtDNA genomes, which is obviously really useful since it’s so fast. In contrast, the local alignment algorithm takes 1 hour to find the nearest neighbor of an input genome over the same dataset. This is obviously much less useful for discovery purposes, but my plan is to use it as further evidence for the histories I uncovered using mostly autonomous, extremely fast global alignment methods. Upon reflection, the gist is, global alignment methods are so fast, they allow for high volume, autonomous discovery, that can then be more carefully considered using local alignments.
I noticed a while back that the purported Quantum Mechanical explanations for the Double-Slit Experiment, don’t make any sense for a very simple reason: generally, people assume the velocity of light is fixed at c. In contrast, if we assume self-interference for a photon, then the photon must (i) change direction or (ii) change velocity, or both, otherwise, there won’t be any measurable effects of self-interference, barring even more exotic assumptions.
A diagram of the Double-Slit Experiment. Image courtesy of Wikipedia.
We can rule out case (ii), since photons are generally assumed to travel at a constant velocity of c in a vacuum. However, case (i) produces exactly the same problem, since if the photon changes direction due to self-interference, then the arrival time against the screen will imply a velocity of less than c, since by definition, the photon will not have followed a straight path from the gun to the screen. This is the end of the story, and I cannot believe that no one has pointed this out before.
We can instead posit a simple explanation for the purported interference pattern: the photons scatter inside the slit (i.e., bounce around), which from our perspective is flat, but from the perspective of a photon, is instead a tunnel, since the photon (or other elementary particle) is so small. This will produce two distributions of scattering angles (one for each slit), which will overlap at certain places along the screen more than others, producing a distribution of impact points that would otherwise look like a wave interference pattern.
This is much simpler isn’t it, and clearly a better explanation than some exotic nonsense about time and other realities. That’s not how you do science. Now, it could be that there is some experiment that requires such exotic theories, but I don’t know of one, not even the Quantum Eraser Experiment. All of these experiments have simple explanations, and we’ve developed a horrible, unscientific habit, of embracing exotic explanations for basic phenomena, that is at this point suspicious, given the military and economic value of science.
I don’t remember the first time I heard about the Liar’s Paradox, but it was definitely in college, because it came up in my computer theory classes in discussions on formal grammars. As such, I’ve been thinking about it on and off, for about 20 years. Wikipedia says that the first correct articulation of the Liar’s Paradox is attributed to a Greek philosopher named Eubulides, who stated it as follows: “A man says that he is lying. Is what he says true or false?” For purposes of this note, I’m going to distill that to the modern formulation of, “This statement is false.”
As an initial matter, we must accept that not all statements are capable of meaningful truth values. For example, “Shoe”. This is just a word, that does not carry any intrinsic truth value, nor is there any meaningful mechanical process that I can apply to the statement to produce a truth value. Contrast this with, “A AND B”, where we know that A = “true” and B = “false”, given the typical boolean “AND” operator. There is in this case a mechanical process that can be applied to the statement, producing the output “false”. Now, all of that said, there is nothing preventing us from concocting a lookup table where, e.g., the statement “Shoe” is assigned the value “true”.
Now consider the distilled Liar’s Paradox again: “This statement is false”. There is no general, mechanical process that will evaluate such a statement. However, it is plainly capable of producing a truth value, since it simply asserts one for itself, much like a lookup table. Typically, this is introduced as producing a paradox, because if we assume the truth value is false, then the truth value of false is consistent with the truth value asserted in the statement. Generally speaking, when assertion and observation are consistent, we say the assertion is true, and this is an instance of that. As such, the statement is true, despite the fact that the statement itself asserts that it is false. Hence, the famous paradox.
Now instead approach the problem from the perspective of solving for the truth value, rather than asserting the truth value. This would look something like, “This statement is A”, where . Now we can consider the two possible values of . If , then the statement asserts a truth value that is consistent with the assumed truth value, and there’s nothing wrong with that. If instead , then we have a contradiction, as noted above. Typically, when engaging in mathematics, contradictions are used to rule out possibilities. Applying that principle in this case yields the result that , which resolves the paradox.
In summary, not all statements are capable of mechanical evaluation, and only a subset of those mechanically evaluable statements resolve to true or false. This however does not prevent us from simply assigning a truth value to a statement, whether by a lookup table, or within the statement itself. However, if we do so, we can nonetheless apply basic principles of logic and mathematics, and if we adhere to them, we can exclude certain purported truth values that are the result of mere assertion. In this case, such a process implies that a statement that asserts its own truth value, is always true.
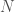 artifacts, there will be
artifacts, there will be  possible rankings. As a consequence, two people discussing, e.g., just 10 works of art, creates 9,765,625 possible rankings, and again, all they have to say is, I hate it, I don’t like it, it’s OK, I like it, or I love it, 10 times, and they’ll convey about 23 binary bits of information, which is
possible rankings. As a consequence, two people discussing, e.g., just 10 works of art, creates 9,765,625 possible rankings, and again, all they have to say is, I hate it, I don’t like it, it’s OK, I like it, or I love it, 10 times, and they’ll convey about 23 binary bits of information, which is  genetic bases worth of information. As you can see, increasing the number of possible rankings simply increases the base of the exponent, whereas the exponent dominates the counting function.
genetic bases worth of information. As you can see, increasing the number of possible rankings simply increases the base of the exponent, whereas the exponent dominates the counting function. possible combinations of artifacts (i.e., the cardinality of the set of all subsets of artifacts), each of which is capable of an ordinal ranking, and therefore, we have, for any
possible combinations of artifacts (i.e., the cardinality of the set of all subsets of artifacts), each of which is capable of an ordinal ranking, and therefore, we have, for any  possible rankings over the set of all subsets of those artifacts. If we set
possible rankings over the set of all subsets of those artifacts. If we set  as we did above, i.e., considering just 10 artifacts, we find that
as we did above, i.e., considering just 10 artifacts, we find that  , which is approximately 1187 genetic bases worth of information, or approximately 7.0% of an entire human mtDNA genome. Now we’re talking about a significant amount of information, that can realistically be conveyed, since
, which is approximately 1187 genetic bases worth of information, or approximately 7.0% of an entire human mtDNA genome. Now we’re talking about a significant amount of information, that can realistically be conveyed, since  , and most people should be able to meaningfully consider around 1000 collections of artifacts, at least over a significant period of time. Just imagine selecting an outfit to wear to a particular gallery exhibition, this is exactly the kind of combination of preferences that we’re considering, which quickly start to cover large numbers of possible combinations. In fact, even having strong preferences of this sort could demonstrate genetic fitness, in addition to conveying genetic information. The net point being, because it’s possible, and it’s obviously something many people take very seriously, it should have some biological function, and I think it’s the obvious: you’re conveying information about your genome through your preferences.
, and most people should be able to meaningfully consider around 1000 collections of artifacts, at least over a significant period of time. Just imagine selecting an outfit to wear to a particular gallery exhibition, this is exactly the kind of combination of preferences that we’re considering, which quickly start to cover large numbers of possible combinations. In fact, even having strong preferences of this sort could demonstrate genetic fitness, in addition to conveying genetic information. The net point being, because it’s possible, and it’s obviously something many people take very seriously, it should have some biological function, and I think it’s the obvious: you’re conveying information about your genome through your preferences. columns, each column representing a base of the genome stored in that row (i.e., each column entry is one of the bases A, C, G, or T, though there are some missing bases, represented by 0’s). Said otherwise, each genome contains
columns, each column representing a base of the genome stored in that row (i.e., each column entry is one of the bases A, C, G, or T, though there are some missing bases, represented by 0’s). Said otherwise, each genome contains 
 and
and  , the number of matching bases is simply 2. Because mtDNA is circular, it’s not clear where to start the comparison. For example, we could start reading genome
, the number of matching bases is simply 2. Because mtDNA is circular, it’s not clear where to start the comparison. For example, we could start reading genome 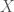 at the first
at the first 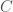 , rather than the first base
, rather than the first base 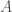 . However, the previous link demonstrates that there’s exactly one whole-genome alignment (otherwise known as a global alignment), or starting index for mtDNA, the rest of them are simply not credible for the reasons discussed above.
. However, the previous link demonstrates that there’s exactly one whole-genome alignment (otherwise known as a global alignment), or starting index for mtDNA, the rest of them are simply not credible for the reasons discussed above. , i.e., 90% of the genome, and retrieve all genomes that are at least a 90% match to Heidelbergensis. This produces the chart below, where the height of the bar provides the percentage of genomes in the applicable population that are at least a 90% match to the single Heidelbergensis genome. For example, 100% of the Iberian Romani are at least a 90% match to the Heidelbergensis genome, producing a height of 1.0 in the chart below. The population acronyms can be found at the back of [2], but just to highlight some of the obvious matches, KZ stands for Kazakhstan, IB stands for Iberian Romani, IT stands for Italy, and RU stands for Russia.
, i.e., 90% of the genome, and retrieve all genomes that are at least a 90% match to Heidelbergensis. This produces the chart below, where the height of the bar provides the percentage of genomes in the applicable population that are at least a 90% match to the single Heidelbergensis genome. For example, 100% of the Iberian Romani are at least a 90% match to the Heidelbergensis genome, producing a height of 1.0 in the chart below. The population acronyms can be found at the back of [2], but just to highlight some of the obvious matches, KZ stands for Kazakhstan, IB stands for Iberian Romani, IT stands for Italy, and RU stands for Russia.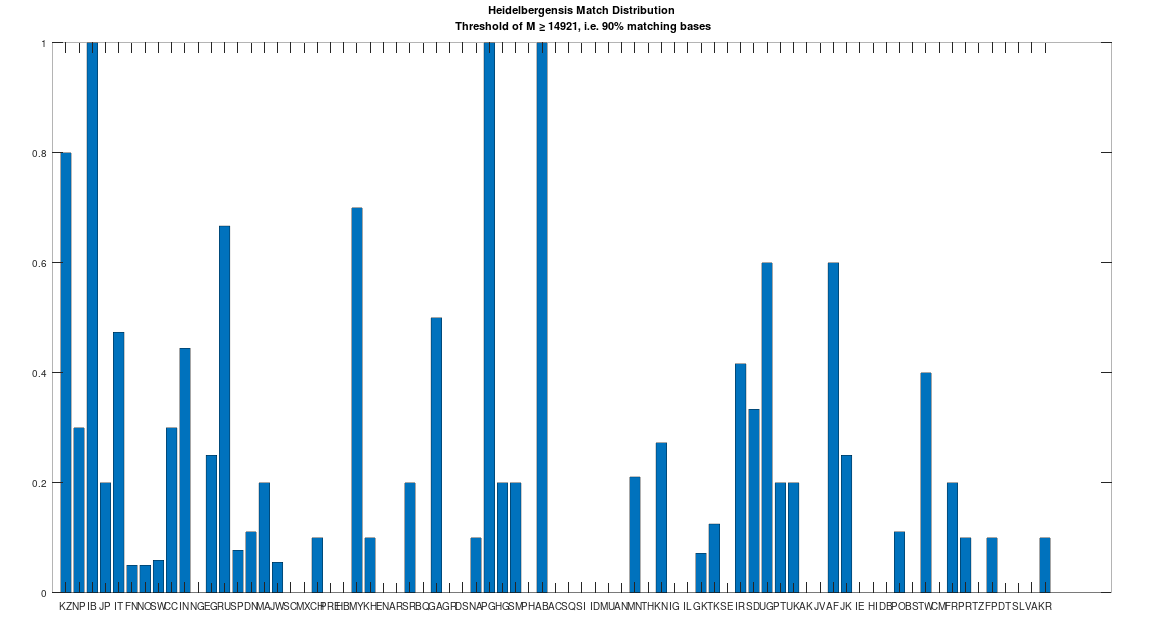
 . That is, at each base, we have two random variables, one for each genome, and each of those variables can take on a value of A, C, G, or T. If it’s truly random, then there are
. That is, at each base, we have two random variables, one for each genome, and each of those variables can take on a value of A, C, G, or T. If it’s truly random, then there are  possible outcomes, and only 4 of those outcomes correspond to the bases being the same, producing a probability of
possible outcomes, and only 4 of those outcomes correspond to the bases being the same, producing a probability of 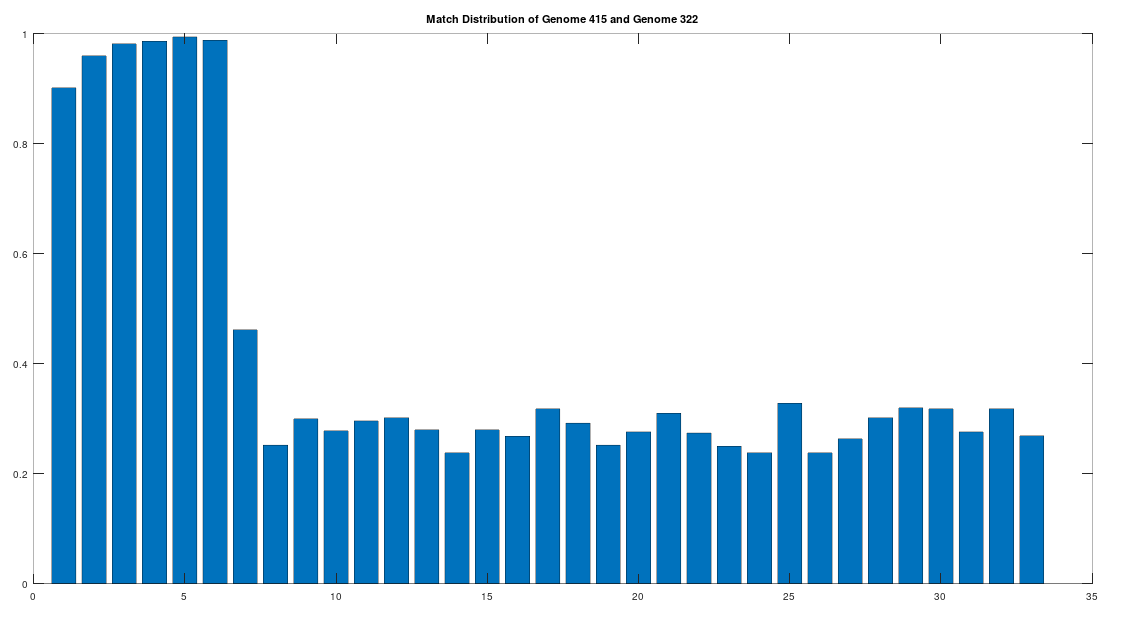



 and
and  . These two genomes have a match count of 3 bases, or 75% of the genome. Now let’s say we create an indel in genome
. These two genomes have a match count of 3 bases, or 75% of the genome. Now let’s say we create an indel in genome  , inserting a “G” after the first “A”, producing the genome
, inserting a “G” after the first “A”, producing the genome  . The match count is now instead 1 base, or 25% of the genome, depending on which genome’s length you use (i.e., 4 or 5).
. The match count is now instead 1 base, or 25% of the genome, depending on which genome’s length you use (i.e., 4 or 5). , a local alignment could, e.g., take the segment
, a local alignment could, e.g., take the segment  from
from  in genome
in genome  mutations occurred over the entire history outlined above, which is not that many, since it spans around 500,000 years. Further, the local alignment algorithm found only 2 indels between the two genomes. This is all consistent with the extremely slow rate of mutation of mtDNA generally. That said, note that unlike the whole-genome algorithm, the local alignment algorithm is approximate, since there is (to my knowledge) no single segment length (in this case 500 bases) that is an objective invariant for comparing two genomes. Said otherwise, when using whole-genome comparison, both mathematical theory and empiricism show there’s only one global alignment, and therefore only one algorithm that gets the job done. In contrast, local alignments can produce different results if we vary the segment length, which is again in this case set to 500 bases. But the bottom line is, there really isn’t that much change over a huge period of time.
mutations occurred over the entire history outlined above, which is not that many, since it spans around 500,000 years. Further, the local alignment algorithm found only 2 indels between the two genomes. This is all consistent with the extremely slow rate of mutation of mtDNA generally. That said, note that unlike the whole-genome algorithm, the local alignment algorithm is approximate, since there is (to my knowledge) no single segment length (in this case 500 bases) that is an objective invariant for comparing two genomes. Said otherwise, when using whole-genome comparison, both mathematical theory and empiricism show there’s only one global alignment, and therefore only one algorithm that gets the job done. In contrast, local alignments can produce different results if we vary the segment length, which is again in this case set to 500 bases. But the bottom line is, there really isn’t that much change over a huge period of time. and
and  , respectively. Let’s further assume that we are surrounded almost exclusively by positive mass, which again is denoted by
, respectively. Let’s further assume that we are surrounded almost exclusively by positive mass, which again is denoted by  to represent a photon, we could write that
to represent a photon, we could write that  , to represent a positron and electron colliding to form a photon. Similarly, we could write that
, to represent a positron and electron colliding to form a photon. Similarly, we could write that  to represent a light on light collision producing an electron-positron pair.
to represent a light on light collision producing an electron-positron pair. and
and  . The question is, what do we get when we combine the negative-mass versions of an electron and positron? Expressed symbolically,
. The question is, what do we get when we combine the negative-mass versions of an electron and positron? Expressed symbolically,  , where
, where  , as a particle distinct from ordinary light. Specifically, if we further posit that negative-mass light does not interact with positive mass, and that positive-mass light (our ordinary photons) do not interact with negative mass, then that negative mass would not be detectable using ordinary photons. Ironically, this is a kind of “dark” matter, but it’s not the nonsense hypothesized hitherto.
, as a particle distinct from ordinary light. Specifically, if we further posit that negative-mass light does not interact with positive mass, and that positive-mass light (our ordinary photons) do not interact with negative mass, then that negative mass would not be detectable using ordinary photons. Ironically, this is a kind of “dark” matter, but it’s not the nonsense hypothesized hitherto. and
and  . If we count matching bases using the first index of each genome, then the number of matching bases is zero. If instead we start at the first index of
. If we count matching bases using the first index of each genome, then the number of matching bases is zero. If instead we start at the first index of  and the second index of
and the second index of  (and loop back around to the first ‘G’ of
(and loop back around to the first ‘G’ of  and
and  . First you’ll note these genomes are not the same length, unlike in the example above, which is another factor to be considered when developing an alignment for comparison. If we simply use the first three bases of each genome for comparison, then the match count will be one, since the first two initial ‘A’s match. If instead we use index two of
. First you’ll note these genomes are not the same length, unlike in the example above, which is another factor to be considered when developing an alignment for comparison. If we simply use the first three bases of each genome for comparison, then the match count will be one, since the first two initial ‘A’s match. If instead we use index two of 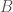 , then the entire
, then the entire  sequence matches, and the resultant match count will be three.
sequence matches, and the resultant match count will be three. . As for accuracy, using Nearest Neighbor to predict the ethnicity of each individual in my dataset produces an accuracy of 30.87%, and because there are 75 global ethnicities, chance implies an accuracy of
. As for accuracy, using Nearest Neighbor to predict the ethnicity of each individual in my dataset produces an accuracy of 30.87%, and because there are 75 global ethnicities, chance implies an accuracy of  . As such, we can conclude that the Nearest Neighbor algorithm is not producing random results, and more generally, produces results that provide meaningful information about the ethnicities of individuals based solely upon their mtDNA, which is remarkable, since ethnicity is a complex trait, that clearly should depend upon paternal ancestry as well.
. As such, we can conclude that the Nearest Neighbor algorithm is not producing random results, and more generally, produces results that provide meaningful information about the ethnicities of individuals based solely upon their mtDNA, which is remarkable, since ethnicity is a complex trait, that clearly should depend upon paternal ancestry as well. of Norwegian genomes map to Pashtuns, who are
of Norwegian genomes map to Pashtuns, who are 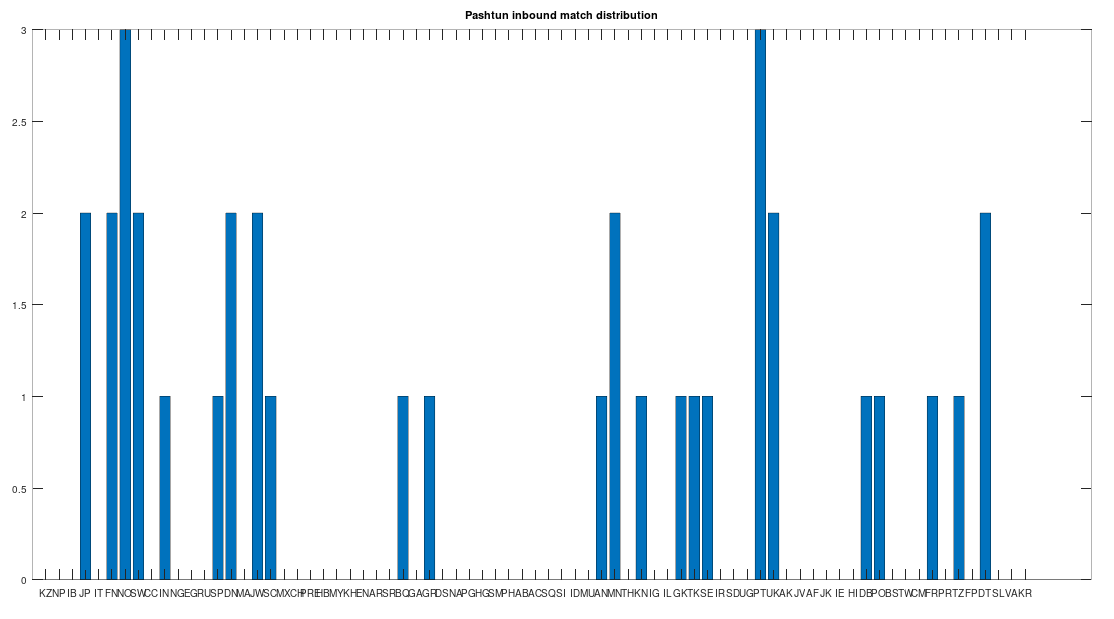
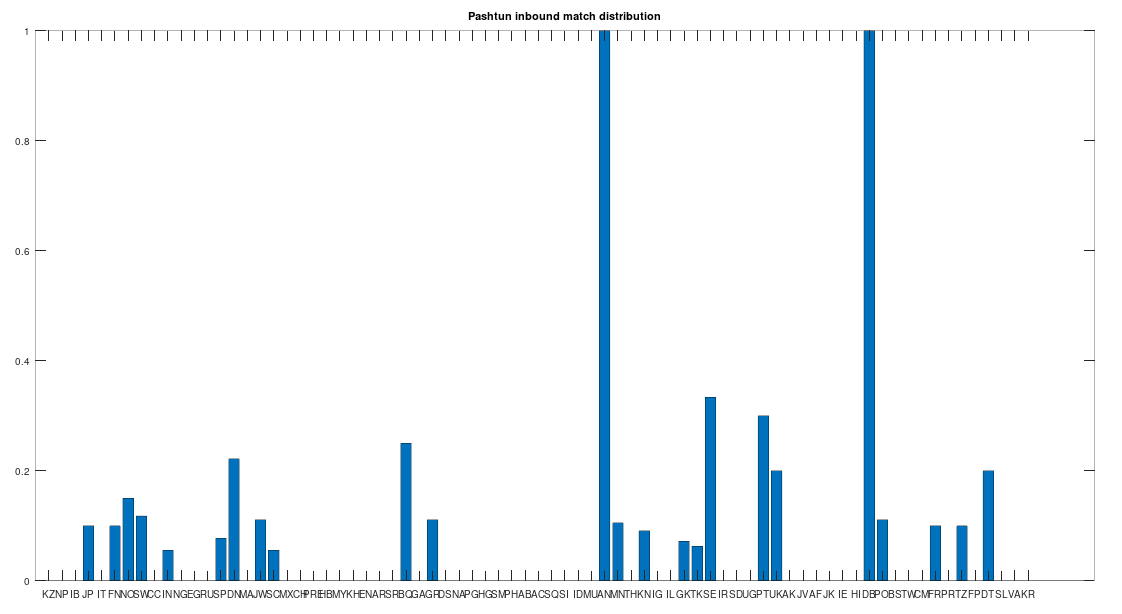
 , so these are plainly meaningful, nearly identical matches. Finally, these Pashtun genomes that are turning up as Nearest Neighbors are heterogeneous. That is, it’s not the case that a single Pashtun genome is popping up globally, and instead, multiple distinct Pashtun genomes are popping up globally as Nearest Neighbors. One not-so-plausible explanation that I think should be addressed is the
, so these are plainly meaningful, nearly identical matches. Finally, these Pashtun genomes that are turning up as Nearest Neighbors are heterogeneous. That is, it’s not the case that a single Pashtun genome is popping up globally, and instead, multiple distinct Pashtun genomes are popping up globally as Nearest Neighbors. One not-so-plausible explanation that I think should be addressed is the 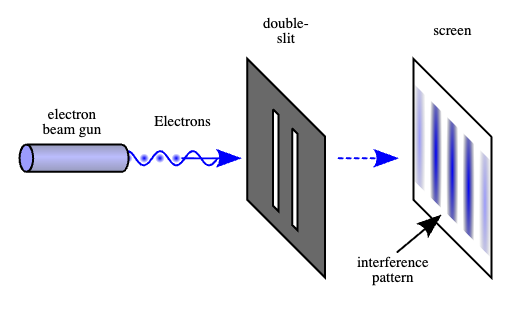
 . Now we can consider the two possible values of
. Now we can consider the two possible values of  , then the statement asserts a truth value that is consistent with the assumed truth value, and there’s nothing wrong with that. If instead
, then the statement asserts a truth value that is consistent with the assumed truth value, and there’s nothing wrong with that. If instead  , then we have a contradiction, as noted above. Typically, when engaging in mathematics, contradictions are used to rule out possibilities. Applying that principle in this case yields the result that
, then we have a contradiction, as noted above. Typically, when engaging in mathematics, contradictions are used to rule out possibilities. Applying that principle in this case yields the result that 
{kind=link}
{kind=link}