My work on mtDNA has led to a thesis that human life begins in Africa, spreads to Asia, and then spreads (1) back West to Europe and Africa and (2) further East into East Asia and the Pacific. I call this the Migration-Back Hypothesis, and you can read all about it here [1], and here, and on my blog generally, where you’ll find a ton of material on topic.
One of the most interesting observations in my work is that the living modern day people of Cameroon test as having the most ancient genomes in the dataset of complete human mtDNA genomes I’ve assembled, which contains 19 archaic mtDNA genomes, that are Heidelbergensis (1 genome), Neanderthal (10 genomes), and Denisovan (8 genomes). This is not too shocking, considering that 53.01% of the 664 genomes in the dataset are at least a 60% match, to at least one archaic genome. This comparison to the archaic genomes is done using the only sensible global alignment for mtDNA, so you can’t argue that it’s chance, or cherry picking, there are a lot of living people that have archaic mtDNA. The reason I’m writing this note is because I think two of the Neanderthal genomes were misclassified by the scientists that sequenced the genomes.
I’ve written previously that the Neanderthals are decidedly heterogenous on the maternal line, in that there are 10 Neanderthal genomes, that can be broken into 6 completely distinct clusters (i.e., groups of similar genomes). I’m using a global alignment for all of this work, except where noted below, and as noted above, there’s only 1 sensible global alignment for mtDNA, so these distinctions are objective.
Specifically, (i) genomes 1, 2, and 10 are at least a 99.5% mutual match to each other, (ii) genomes 5 and 6 are a 63.4% match to each other, (iii) genomes 8 and 9 are a 99.9% match to each other, and (iv) genomes 3, 4, and 7 are unique, and have no meaningful match to each other or the rest of the Neanderthal genomes. This note focuses on genomes 5 and 6, which appear to be misclassified as Neanderthals, and instead seem to be Denisovans based upon their mtDNA. All of the provenance files for the relevant genomes are linked to below at the bottom of the article, and each provenance file includes a FASTA file that contains the applicable full genome. The full dataset I’ve assembled (which includes all of these archaic genomes) is available in [1] above.
Neanderthal Genome 5
The provenance file for Neanderthal Genome 5 (row 389 of my dataset) lists the “organism” field as “Homo sapiens neanderthalensis”, and the “sub_species” field as “neanderthalensis”. However, the genome title includes the phrase “Denisova 17″, and the “isolate” field is listed as “Denisovan 17”. Further, the article associated with the genome suggests that the genome is actually from the Denisova Cave in Siberia, yet they classified it as Neanderthal, which doesn’t look right. The relevant quote is on page 30 (page 3 of the pdf):
We estimated the molecular age of the mtDNA of the newly identified Neanderthal (Denisova 17) to ~134 ka (95% height posterior density (HPD): 94–177 ka) using Bayesian dating…
Note that “Denisovan 17” is a label used by the authors of the quoted article, I’m using indexes and row numbers keyed to my dataset (i.e., “Denisovan 17” is Neanderthal Genome 5 in my dataset). However, as noted above, Neanderthal Genome 5 is a 63.4% match to Neanderthal Genome 6 only, and is not a significant match to any other Neanderthal genome. This suggests that these two genomes are, as noted above, a distinct maternal line that lived among other maternal lines, that have all been archeologically classified as Neanderthals. However, Neanderthal Genome 5 was found in the Denisovan Cave in Siberia, per the article quoted above, which is already evidence for the claim that it is actually a Denisovan, at least with respect to its maternal line.
Further, Neanderthal Genome 5 has 8,915 bases (i.e., 53.77% of the full genome) in common with Denisovan Genome 1 (row 377 of my dataset), using the whole genome global alignment, which is well beyond chance (i.e., 25.00% of the full genome). In contrast, Neanderthal Genome 5 has 5,300 bases (i.e., 31.96% of the full genome) in common with its closest match among the other Neanderthal Genomes (save for Neanderthal Genome 6, which also seems to be Denisovan, and is discussed below).
Finally, Neanderthal Genome 5 has 16,328 bases (i.e., 98.48% of the full genome) in common with a Cameroon Genome (row 591 of my dataset). That Cameroon Genome in turn has 8,898 bases (i.e., 53.47% of the full genome) in common with the same Denisovan Genome 1 (row 377 of my dataset). The plain conclusion is that Neanderthal Genome 5 is an archaic Siberian Denisovan individual, with a close maternal connection to living West Africans. As noted above, the Cameroon test as the most ancient people across my dataset, suggesting a migration from Cameroon to Siberia, which is consistent with the Out of Africa Hypothesis, but does not contradict my Migration-Back Hypothesis, since it’s entirely possible that later Denisovans migrated back to Europe or Africa from Siberia, or further into East Asia and the Pacific. However, that is not the point of this note, which is limited to the misclassification of two Neanderthal genomes.
Neanderthal Genome 6
Similarly, Neanderthal Genome 6 has 5,289 bases (i.e., 31.90% of the full genome) in common with its closest match among the other Neanderthal Genomes (save for Neanderthal Genome 6, which also seems to be Denisovan, as discussed above). In contrast, Neanderthal Genome 6 has 8,588 bases (i.e., 51.80% of the full genome) in common with Denisovan Genome 1 (row 377 of my dataset). Further, Neanderthal Genome 6 has 10,461 bases (i.e., 63.09% of the full genome) in common with the same Cameroon genome discussed above. However, unlike Neanderthal Genome 5, the provenance file for Neanderthal Genome 6, and the related article, make it clear the genome was discovered in Scladina, which is an archeological site in Belgium. Even using a local alignment, the resultant number of matching bases between Neanderthal Genome 6 and the Cameroon genome is 16,183, which is lower than the number of matching bases between Neanderthal Genome 6 and that same genome (i.e., 16,328) using a global alignment. Note that local alignments maximize the number of matching bases. The sensible conclusion being that Neanderthal Genome 6 is actually Denisovan, though it is not as close to the Cameroon genome as Neanderthal Genome 5, though it is close enough to infer African ancestry. This is again consistent with the Out of Africa Hypothesis, though it’s not clear whether this genome has any connection to Asia, at least limited to this discussion alone, and as such, it adds no further credibility to my Migration-Back Hypothesis, though it does not contradict the Migration-Back Hypothesis in any way, since it’s entirely possible at least some people left Africa directly for Europe or other places. In contrast, the Migration-Back Hypothesis is about the overall migration patterns of some of the most modern mtDNA genomes in the dataset, linking otherwise disparate modern humans across enormous distances.
In a paper entitled, “A New Model of Computational Genomics” [1], I introduced an algorithmic test for ancestry using whole-genome mtDNA. I’ve since updated that test significantly, as described below. In this first of what will be a series of articles, I will present the results of this test as applied to specific regions of the world, in this case, to Scandinavia. Each of the articles will contain an independent summary of the algorithm and its overall results, and so you can read each independently.
Algorithmic Testing for Ancestry
Assume you’re given whole mtDNA genomes A, B, and C. The goal is to test whether genome A is the ancestor of both genomes B and C. It turns out, this is straight forward as a necessary (but not sufficient condition) for ancestry. Specifically, if we begin with genome A, and then posit that genomes B and C mutated independently away from genome A (e.g., groups B and C travelled to two distinct locations away from group A), then it is almost certainly the case that genomes B and C have fewer bases in common with each other, than they have in common with genome A.
For intuition, because we’ve assumed genomes B and C are mutating independently, the bases that mutate in each of B and C are analogous to two independent coins being tossed. Each mutation will reduce the number of bases in common with genome A. For example, if genome B mutates, then the number of bases that A and B have in common will be reduced. Note we are assuming genome A is static. Because B and C are mutating independently, it’s basically impossible for the number of bases in common between B and C to increase over time. Further, the rate of the decrease in common bases is almost certainly going to be higher between B and C, than between A and B, and A and C. For example, if there are 10 mutations in each of genomes B and C (i.e., a total of 20 mutations combined), then the match counts between A and B and A and C, will both decrease by exactly 10, whereas the match count between B and C should decrease by approximately 20. Let |AB| denote the match count between genomes A and B. We have then the following inequalities:
Case 1: If genome A is the common ancestor of both genomes B and C, then it is almost certainly the case that |AB| > |BC| and |AC| > |BC|. See, [1] for further details.
Even though this is only a necessary condition for ancestry, this pair of inequalities (coupled with a lot of research and other techniques), allowed me to put together a complete, and plausible, history of mankind [2], all the way back to the first humans in Africa.
Ancestry from Archaic Genomes
The simple insight I had, was that if A is not archaic, and B is archaic, then A can’t credibly be the ancestor of B. That is, you can’t plausibly argue that a modern human is the ancestor of some archaic human, absent compelling evidence. Further, it turns out the inequality (since it is a necessary but not sufficient condition) is also consistent with linear ancestry in two cases. Specifically, if |AB| > |BC| and |AC| > |BC|, then we can interpret this as consistent with –
Case 2: B is the ancestor of A, who is in turn the ancestor of C.
Case 3: C is the ancestor of A, who is in turn the ancestor of B.
If you plug in A = Phoenician, B = Heidelbergensis, and C = Ancient Egypt, you’ll find the inequality is satisfied for 100% of the applicable genomes in the dataset. Note that the dataset is linked to in [1]. It turns out you simply cannot tell what direction time is running given the genomes alone (unless there’s some trick I’ve missed), and so all of these claims are subject to falsification, just like science is generally. That said, if you read [2], you’ll see fairly compelling arguments consistent with common sense, that Heidelbergensis (which is an archaic human), is the ancestor of the Phoenicians, who are in turn the ancestors of the Ancient Egyptians. This is consistent with case (2) above.
Putting it all together, we have a powerful necessary condition that is consistent with ancestry, but not a sufficient condition, and it is therefore subject to falsification. However, one of these three cases is almost certainly true, if the inequalities are satisfied. The only question is which one, and as far as I can tell, you cannot determine which case is true, without exogenous information (e.g., Heidelbergensis is known to be at least 500,000 years old). You’ll note that cases (1), (2), and (3) together imply that A is always the ancestor of either B or C, or both. My initial mistake was to simply set B to an archaic genome, and assert that since A cannot credibly be the ancestor of B, it must be the case that A is the ancestor of C. Note that because A cannot credibly be the ancestor of B, Cases (1) and (3) are eliminated, leaving Case (2), which makes perfect sense: B is archaic, and is the ancestor of A, who is in turn the ancestor of C. However, this is not credible if C is also archaic, producing a lot of bad data.
Updated Ancestry Algorithm
The updated algorithm first tests literally every genome in the dataset, and asks whether it is at least a 60% match to an archaic genome, and if so, it treats that genome as archaic for purposes of the test, so that we avoid the problem highlighted above. This will allow us to reasonably assert that all tests involve exactly one archaic genome B, and therefore, we must be in Case (2). Interestingly, some archaic populations were certainly heterogenous, which is something I discussed previously. As a result, there are three ostensibly archaic genomes in the dataset, that do not match to any other archaic genomes in the dataset, and they are therefore, not treated as archaic, despite their archeological classification. You can fuss with this, but it’s just three genomes out of 664, and a total of 19,972,464 comparisons. So it’s possible it moved the needle in marginal cases, but the overall conclusions reached in [2] are plainly correct, given the data this new ancestry test produced.
There is however the problem that the dataset contains only Heidelbergensis, Denisovan, and Neanderthal genomes, leaving out e.g., Homo Erectus, and potentially other unknown archaic humans. There’s nothing we can do about this, since we’re constantly finding new archaic humans. For example, Denisovans were discovered in 2010, which is pretty recent, compared to Heidelbergensis, which was discovered in 1908. Moreover, the three genomes in question are possibly three new species, since they don’t match to Denisovan, Heidelbergensis, or Neanderthals. All of that said, taken as a whole, the results produced by this new algorithm, which makes perfect theoretical sense and must be true, are consistent with the results presented in [2]. Specifically, that humans began in Africa, somewhere around present day Cameroon, migrated to the Middle East, then Asia, producing the three most evolved maternal lines that I’ve identified, somewhere around Nepal, specifically, the Ancient Egyptians, the Vikings, and the Ancient Romans. The first two maternal lines are both found around the world, and descend from Heidelbergensis and Neanderthals and / or Denisovans, respectively, suggesting that many modern humans are a mix between the most evolved maternal lines that originated in three distinct archaic human populations, effectively creating hybrids. The Ancient Roman maternal line no longer exists, and seems to have been deliberately annihilated. For your reference, you can search for the Pre Roman Ancient Egyptian genome (row 320, which descends from Heidelbergensis) and the Icelandic genome (row 464, which descends from either Neanderthals or Denisovans, or both, it’s not clear).
Maternal Ancestry Among Scandinavians and Germans
Intuition suggests that the Sami People, who are indigenous Scandinavians, should as a general matter test as the ancestors of at least some Scandinavian people. At the same time, because all but the Finns and Sami speak Germanic languages, we would expect the Germans to test as the ancestors of at least some Scandinavian people. All of that said, during the Viking Age, the Scandinavians made use of a Phoenician-like alphabet, known as Runes, and so it’s at least possible we should see either Continental European ancestry (e.g., the Basque used similar scripts in antiquity), Middle Eastern ancestry, or some other form of ancestry that explains this otherwise anomalous alphabet. We will examine each of these questions below using the ancestry test.
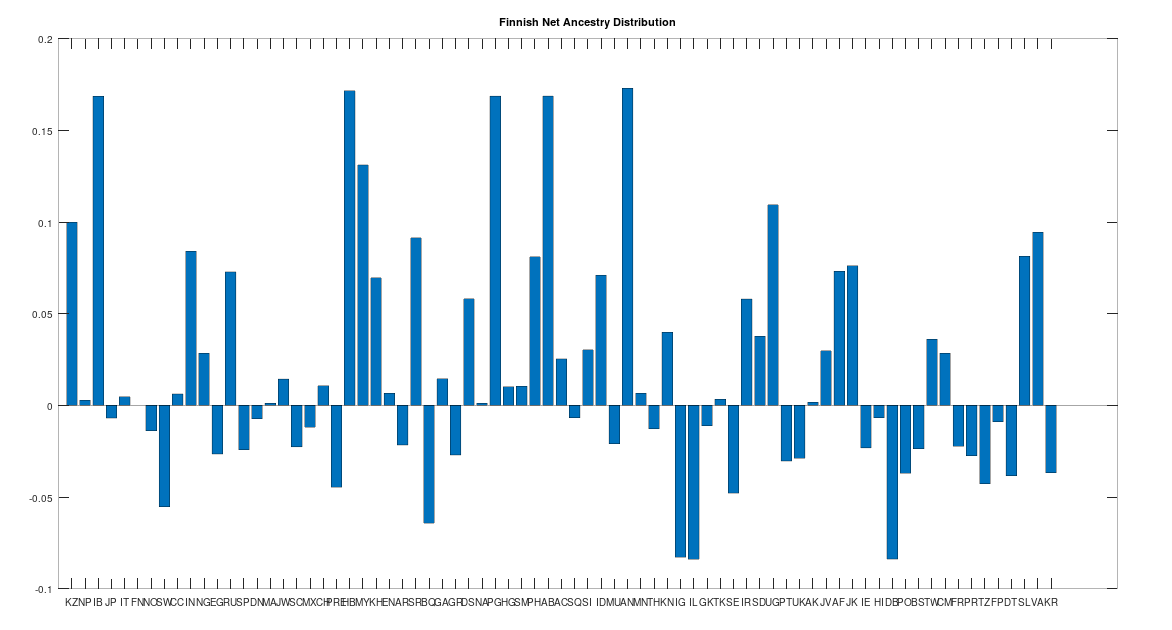
Levänluhta
Levänluhta is an underwater gravesite in Finland that contains the remains of about 100 individuals from the Iron Age (c. 800 to 500 BC). Though Scandinavia has been occupied by humans since the Stone Age, common sense says that these individuals should test as the ancestor of at least some modern Scandinavians. This is indeed the case, and in fact, these individuals test as even more ancient than the Sami People, which you can see in the chart below. A positive number indicates that the population in question is a net ancestor, whereas a negative number indicates that the population in question is a net descendant. That is, if e.g., X is the number of times the ancestry test was satisfied from Sweden to Norway, and Y is the number of times the ancestry test was satisfied from Norway to Sweden, the chart below plots X – Y for each population. As you can see, all other Scandinavian groups test as the descendants of the individuals buried in Levänluhta. You can find the acronyms used below at the end of [1], but for now note that FN = Finland, NO = Norway, SW = Sweden, DN = Denmark, SM = Sami, IL = Iceland, and AF = Ancient Finland (i.e., Levänluhta). If you look at the ancestors of the individuals buried in Levänluhta (i.e., X – Y > 0), you’ll see HB = Heidelbergensis, AN = Andamanese, and other archaic populations, suggesting the individuals buried in Levänluhta are somewhere between archaic humans and modern humans, despite being a relatively recent Iron Age gravesite.
The Sami People
The Sami People are indigenous Scandinavians that speak an Uralic language and live in Northern Scandinavia, spanning Sweden, Norway, Finland, and Russia. For context, Uralic languages are spoken in regions around Finland, including Finland itself, Estonia, parts of Russia, as well Hungary. Uralic languages are to my knowledge not related to Germanic languages. As such, we should not be surprised if the Sami have a maternal ancestry that is distinct from the rest of the Scandinavians and Germans. This is in fact the case, and in particular, the Sami contain a significant amount of Denisovan mtDNA. See, [1] for more details. As noted above, Denisovans are a relatively recently discovered subspecies of archaic humans. The main archeological site where they were discovered is the Denisovan Cave in Siberia, and the dataset includes 8 Denisovan genomes from that site.
Above is the net maternal ancestry of the Sami people, where, again, a positive number indicates that the population in question is an ancestor of the Sami, and a negative number indicates that the population in question is a descendant of the Sami. As you can see above, all other living Scandinavian people test as the descendants of the Sami, making the Sami the most ancient among the living Scandinavian people.
The Finnish People
As noted above, the Finnish people speak an Uralic language, like the Sami, and as such, we should not be surprised if they have a distinct ancestry from the rest of the Scandinavians. This is in fact the case, though they are one step closer to modern Scandinavians than the Sami, and as you can see below, all Scandinavian people (other than the Sami) test as the descendants of the Finns.
Now this doesn’t mean that all the other Scandinavians descend directly from the Finns, which is too simple of a story, but it does mean that when comparing Finns to the rest of the Scandinavians (save for the Sami), it is more likely that a given Finn will test as the ancestor of a given Scandinavian, than the other way around. This is not terribly surprising since the Finns speak a completely different language that has (to my knowledge) an unknown origin, suggesting the language is quite ancient, and the Finns seem to be as well. The Finns also have a significant amount of Denisovan mtDNA from Siberia, which is again consistent with the claim that the Finns are, generally speaking, the second most ancient of the living Scandinavians.
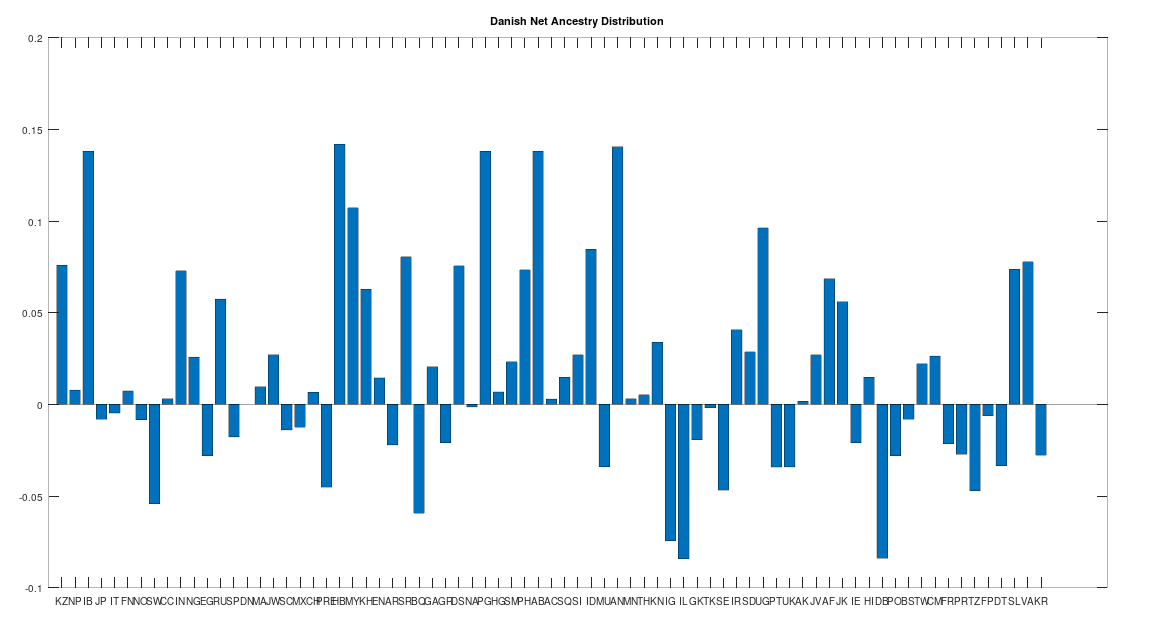
The Danish People
Like the Finns, the Danes also contain a significant but lesser amount of Siberian Denisovan mtDNA, and they similarly test as the ancestors of all other Scandinavians, other than the Finns and Sami, making them the third most ancient Scandinavian population. Note however that Danish is a Germanic language, suggesting independence between Uralic languages and Denisovan mtDNA, though there does seem to be some reasonable correlation.
The Norwegian People
The Norwegian people contain no meaningful quantity of Denisovan mtDNA, and they test as the fourth most ancient of the living Scandinavians. Note that the Sami, Finns, and Danes test as the net ancestors of the Norwegians, whereas the Swedes and Icelandic people test as the descendants of the Norwegians. Finally note that the Norwegians speak a Germanic language.
The Swedish People
The Swedes contain no meaningful quantity of Denisovan mtDNA, and they test as the fifth most ancient of the living Scandinavians, and are therefore more modern than the rest, save for the Icelandic (discussed below). The Swedes speak a Germanic language that is very similar to Norwegian, though the Swedes are notably distinct from the Norwegians in that they test as the descendants of the Germans, whereas the rest of the Scandinavians discussed thus far test as the ancestors of the Germans.
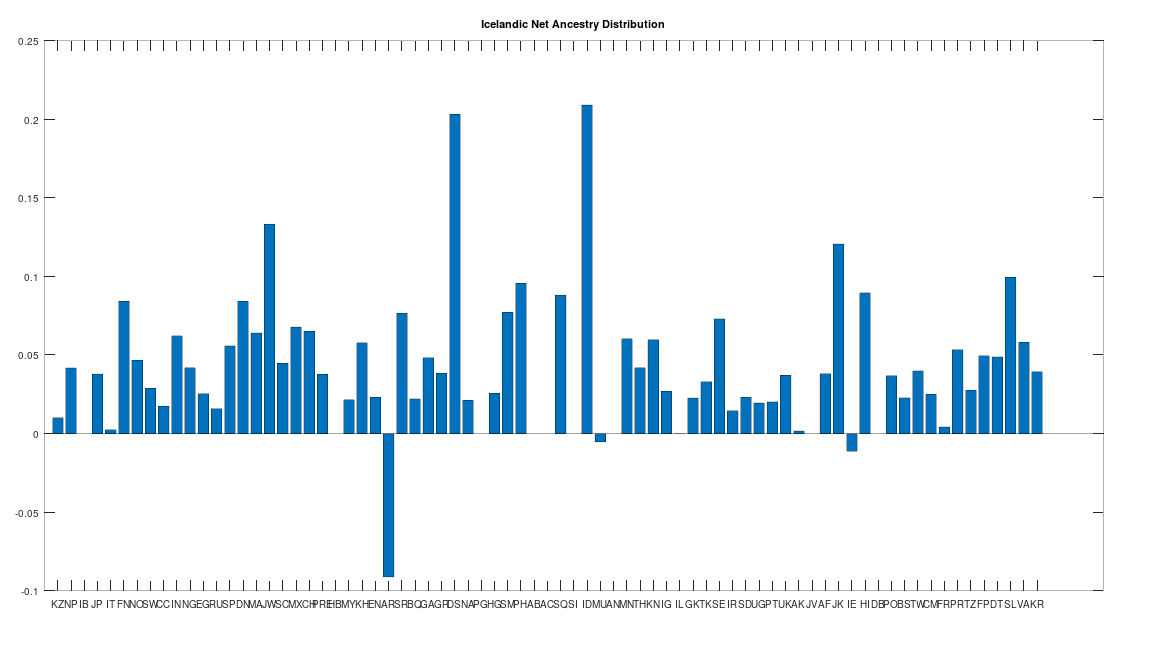
The Icelandic People
There is only one Icelandic genome in the dataset, but as you can see below, it is very similar to the Swedish population generally. Further, this genome tests as the descendant of all Scandinavian populations, and more generally, has only three descendants: the Ancient Romans, the Irish, and the Munda people of India. The Ancient Romans generally test as the descendants of the Northern Europeans, and are in fact the most modern population in the dataset according to this test. The Munda people of India are probably not Scandinavian, and instead, the Scandinavians and the Munda presumably have a common ancestor in Asia, consistent with the “Migration-Back Hypothesis” I presented in [2], that humanity begins in Africa, spreads to Asia, and then back to Northern Europe and Africa, as well as spreading into East Asia. Dublin was founded by the Vikings, so it is no surprise that some Irish test as the descendants of the Icelandic. However, there is only one Icelandic genome in the dataset, and so while we can’t say much about the Icelandic people in general on the basis of the dataset alone, because Iceland was (to my knowledge) uninhabited prior to the Vikings, it’s presumably the case that the people of Iceland are literally direct descendants of the Vikings, whereas in contrast, Scandinavia (as noted above) has been inhabited by humans since the Stone Age.
The Origins of the Runic Alphabet
Note that the Swedes and Icelandic are the only Scandinavians that test as a descendant as opposed to an ancestor of the Germans. This could explain why the majority of the Rune Stones are in Sweden, as opposed to the rest of Scandinavia. Specifically, the hypothesis is that Germanic people brought the Phoenician-like alphabet of the Runic Scripts to Sweden. As noted above, the Basque used a similar alphabet, who are also of course Continental Europeans, and so the overall hypothesis is that people of the Mediterranean (e.g., the Phoenicians themselves) brought their alphabet to the Continental Europeans, and the Germans brought that alphabet to the Swedes.
Asian and African Ancestors and Descendants of the Scandinavians
You’ll note in the charts above that some African and Asian people test as the ancestors and / or the descendants of the Scandinavians, in particular the Nigerians and Tanzanians, and the Koreans, Thai, and Japanese (though there are others). Though this might initially seem puzzling, it is instead perfectly consistent with the Migration-Back Hypothesis presented in [2], which asserts that many modern humans, in particular Northern Europeans, East Asians, and many Africans are the descendants of common ancestors from Asia.
The Ancient Mediterranean
The Ancient Romans are clearly descendants of the Northern Europeans, but I’ve found similar Italian genomes that are 35,000 years old. This implies that the most evolved genomes in the dataset are still at least 35,000 years old, and were already in Italy, long before Ancient Rome. The question is then, if the stage was set 35,000 years ago, in that the modern maternal lines were fully formed, why is that it took so long for civilization to develop? One possibility is that there was further evolution on the male line, or the rest of the genome, which is probably true given that mtDNA is, generally speaking, very slow to evolve.
However, civilization has geography to it, and it is simply impossible to ignore the Mediterranean, which produced the Ancient Egyptians, Mesopotamians, Ancient Greeks, and Ancient Romans, as well as others. Why did these people so drastically outperform literally all other humans? I think the answer is written language, and in turn, mathematics. That is, my hypothesis is that the genetics only gets you so far, and that you’ll find people very similar to e.g., the Phoenicians and Ancient Egyptians in other parts of the world that simply didn’t produce on the scale that the Mediterraneans did, and that the gap was driven by written language, which in turn allows for written mathematics, and everything that follows, from accurate inventories and contracts, to predictions about the future. That said, of all the Ancient and Classical people in the dataset, none of them contain any archaic mtDNA, suggesting maternal evolution really did play a role in intelligence and human progress.
This is difficult for modern people to appreciate, but imagine having no idea what happened a few weeks ago, and how that could leave you at a loss, or even put you at risk. At a minimum, written records reduce the risk of a dispute. Now imagine having no written system of mathematics, and trying to plan the construction of a structure, or travel over a long period of time. You’d have no means of calculating the number of days, or the number of individuals required, etc. Once you cross this milestone, it becomes rational to select mates on the basis of intelligence, which is a drastic shift from what happens in nature, which is selection for overall fitness. This seems to create a feedback loop, in that as civilizations become more sophisticated, intelligence becomes more important, further incentivizing selection for intelligence, thereby creating a more intelligent people.
This is not to diminish the accomplishments of other people, but it’s probably the case that the Mediterranean people of the Ancient and Classical periods were the most intelligent people in the world, at the time, which forces the question, of what happened to them? There’s unambiguous evidence that they were literally exterminated, at least in the case of the Romans. The thesis would therefore be that the Romans were slowly and systematically killed to the point of extinction, by less evolved people, creating the societal collapse and poverty that followed for nearly 1,000 years, until the Renaissance.
Unfortunately, it seems plausible the same thing is happening again. Specifically, consider that there have been no significant breakthroughs in physics since Relativity, which we now know is completely wrong. Also consider the fact that the most powerful algorithm in Machine Learning is from 1951. Not surprisingly, microprocessors have been designed using what is basically A.I., since the 1950s. So what is it then that these ostensible A.I. companies do all day? They don’t do anything, it’s impossible, because the topic began and ended in 1951, the only thing that’s changed, is that computers became more powerful. They are with certainty, misleading the public about how advanced A.I. really is, and it’s really strange, because scientists during the 1950s and 1960s, weren’t hiding anything at all. Obfuscation and dishonesty are consistent with a nefarious purpose, and companies like Facebook probably are criminal and even treasonous enterprises, working with our adversaries, and are certainly financed by backwards autocracies like Saudi Arabia.
If you’re too intelligent and educated, then you will know that the modern A.I. market is literally fake, creating an incentive to silence or even kill the most intelligent people, which is consistent with the extremely high suicide rate at MIT. It suggests the possibility that again, intelligent people are being exterminated, and having a look around at the world, it’s obvious that civilization is again declining, arguably when compared to the turn of the 20th Century, and certainly since the end of World War II. I think we all know who’s responsible, and it’s probably not Scandinavians.
In a paper I wrote entitled A New Model of Computational Genomics [1], I presented a simple test for ancestry that is impossible to argue with. Let |AB| denote the number of matching bases between two genomes A and B. Given genomes A, B, and C, if we assume that genome A is the common ancestor of genomes B and C, then it is almost certainly the case (see [1] for a discussion of the probabilities) that |AB| > |BC| and |AC| > |BC|. That is, genomes A and B, and A and C, almost certainly have more bases in common than genomes B and C. For intuition, beginning with genome A, and assuming independent mutations away from A to genomes B and C, this is like tossing two independent coins (i.e., the mutations within genomes B and C that deviate from A), which should not have more than chance in common. As such, B and C should deviate away from each other at a faster rate than they deviate from A individually.
Now this is already really powerful, and led me to a complete history of mankind, which is more than plausible. But that said, it’s a necessary condition, not a sufficient condition. That is, if genome A is the common ancestor of genomes B and C, then the inequalities above almost certainly hold, but it’s subject to falsification (i.e., it’s not a sufficient condition). I realized tonight, you can actually transform this into a necessary and sufficient condition.
Specifically, the inequality above can be represented as a graph where A is connected to B, A is connected to C, and B is connected to C, with the match counts labelling the edges of the graph. For example, the edge connected A to B would be labeled with |AB|, which will be some integer. If the inequalities are satisfied, only two such graphs out of six are plausible, for the same reasons that underly the inequality. Specially, if I assume A is the ancestor of B, which is in turn the ancestor of C, then A and C almost certainly have fewer bases in common than A and B.
The graphs that remain, imply that if the inequality is satisfied, then A is almost certainly the ancestor of either B or C, or both, as a necessary and sufficient condition. If we plug in an implausible genome for either B or C (e.g., assuming that the Norwegians = A are the ancestors of Heidelbergensis = B), then the inequality serves as a necessary and sufficient condition for the descendants of the Norwegians, i.e., genome C. I will write more about this tomorrow, including code and some testing.
UPDATE 10/19/25
I’ve implemented a new version of the ancestry algorithm, which so far seems to work. Code is attached below, more to come!
I noticed a while back that individual subspecies of archaic humans were actually heterogenous, at least with regards to their mtDNA. In particular, the Neanderthal genomes in my dataset are actually 6 completely different maternal lines. There are 10 Neanderthal genomes in total, and the breakdown is (i) genomes 1, 2, and 10 are at least a 99.5% mutual match to each other, (ii) genomes 5 and 6 are a 63.4% match to each other, (iii) genomes 8 and 9 are a 99.9% match to each other, and (iv) genomes 3, 4, and 7 are unique, and have no meaningful match to each other or the rest of the Neanderthal genomes. Further, clusters (i), (ii), and (iii) have no meaningful match to each other. The plain result is that we actually have a heterogenous group of genomes, that have nonetheless been classified as Neanderthal.
Now I’m in no position to criticize archaeological work, but you can’t ignore the fact that we have 6 completely distinct classes of genomes. Because, by definition, there must be 6 distinct maternal lines in this population, it’s probably the case that the rest of the genome also differs meaningfully, though note the number of paternal lines could be larger or smaller than 6. But the point remains, the genomes probably differ generally, not just along the maternal line.
As a result, we have to ask whether we actually have a single subspecies. If we take that view, then the subspecies is the result of the mixing of these 6 distinct maternal lines. And this makes perfect sense, because the vast majority of human populations have heterogeneous maternal lines, and the only exceptions I’m aware of are the Romani People and the Papuans, who are almost perfectly homogenous on the maternal line. It’s worth noting that Romani mtDNA is basically identical to Papuan mtDNA, so there’s probably something to that.
We could instead take the view that the archeological classification is wrong, and that mtDNA controls the definition of a subspecies. I think this is a little aggressive, given that mtDNA is a very small portion of the overall human genome. But at the same time, mtDNA conveys a lot of information about heredity and even conveys information about paternal ancestry, which is amazing. That said, I think the better view is that a given group of people is (generally speaking) the result of a heterogenous group of people that is roughly stable over some period of time, in terms of its distribution of underlying genomes. This apparently applies to archaic humans as well, who seem to be (in at least this case) heterogenous.
Interestingly, the Denisovan genomes in the dataset are all a 97% match to each other, except one, which is totally unique. All of the genomes were (based upon the provenance files) taken from Denisova Cave in Siberia. Though we can’t know, it’s at least possible Denisovans were a more insular group of people than the Neanderthals. It’s possibly unscientific, but the Finns have a lot of Denisovan mtDNA, and they speak a language that is totally different from the Swedes, Norwegians, and Russians, despite sharing large borders with all three countries, suggesting the Finns really are an insular people.
Below are links to the genomes on the NIH website:
In my paper, A New Model of Computational Genomics [1], I introduce a simple test for ancestry that cannot credibly be argued with. The argument is as follows: assume that we begin with genome A in location a, and that three groups of individuals with genome A all begin in location a. Now assume that two of those groups go to different locations, specifically, that one group goes to location b and the other group goes to location c. Because mtDNA is so stable, it could be the case that even over significant amounts of time, the populations in locations b and c, still have genome A, with basically no mutations. If however, any mutations occur, it cannot credibly be the case that genomes in location b (genome B) and location c (genome C) develop even more bases in common with each other. This becomes increasingly unlikely as a function of the number of new matching genomes between B and C, and is governed by the binomial distribution. As a consequence, if A is the common ancestor of genomes B and C, it must be the case that |AB| < |BC| and |AC| < |BC|, where |xy| denotes the number of matching bases between genomes x and y. That is, A must have more bases in common with B and C, than B and C have in common with each other, since B and C independently mutated away from genome A.
Applying this test, we find that the Old Kingdom Ancient Egyptians are the common ancestors of basically all Northern Europeans, many Africans, Asians, and in particular, South East Asians. I’ve also noted repeatedly that the Old Kingdom Ancient Egyptians appear to be Asian, which, superficially, makes no sense. Finally, I’ve noted that Heidelbergensis plainly evolved into Phoenicians, and then the Old Kingdom Ancient Egyptians. Phoenicians appear in Asia on the maternal line, in particular in Sri Lanka.
Putting it all together, tonight I tested which population is most likely to be the ancestor of the Old Kingdom Ancient Egyptians, and the clear answer is the Sri Lankans. The attached code runs the test, and produces a normalized score. The Sri Lankans scored 17.36, and the next best answer was the Vedda Aboriginals (also in Sri Lanka), with a score of 8.3064. The plain implication is that the mutation from the Phoenician maternal line, into the Old Kingdom Ancient Egyptian maternal line took place in Sri Lanka, or somewhere very close.
This completes the history of mankind, with the people of Cameroon the likely source population of all of mankind (including the Denisovans, Heidelbergensis, and Neanderthals), Heidelbergensis then evolving into the Phoenicians, the Phoenicians traveling to Asia, there evolving into the Old Kingdom Ancient Egyptian maternal line, who then migrated back to North East Africa, forming the cradle of modern human mtDNA all over the world, suggesting they were even more successful as a people than current history suggests.
I’ve written in the past on the topic of the rate of mutation of mtDNA, in an attempt to calculate the age of mankind. It turns out, there really isn’t a good single answer to the rate at which human mtDNA mutates, and as a result, you really can’t come to any clear answer using mtDNA alone. And in fact, I realized the other day, that it seems to vary by maternal line. Specifically, some modern humans carry archaic mtDNA, in particular Heidelbergensis, Denisovan, and Neanderthal mtDNA. Other modern humans carry mtDNA that is basically identical to ancient mtDNA (e.g., 4,000 years old), but not archaic mtDNA (e.g., 100,000 years old). In particular, many modern humans globally carry Ancient Egyptian mtDNA, from about 4,000 years ago.
You can get an idea of the rate of mutation, by taking e.g., a modern human that has Denisovan mtDNA, and comparing that to a bona fide archaic Denisovan genome, count the number of changed bases, and then divide by the number of years since the archaic sample lived, which will produce a measure of the number of changed bases per year. This can of course be expressed as a percentage of the total genome size, which is what I’ve done below.
We can be a bit fancier about it, by comparing a given genome to many others, producing a distribution of the number of changed bases per year. The code below does exactly this, producing the average total percentage change, minimum total change, maximum total change, and standard deviation over all total changes. The comparison was made only to modern genomes, and so we can take the known (and plainly approximate) date of the archaic / ancient genome, and divide by the number of years to the present. This will produce a rate of change per year, which I’ve expressed as a rate of change per 1,000 years.
The results are as follows:
GenomeType
Avg. Change
Min. Change
Max. Change
Std. Deviation
Genome Date
Avg. Change Per 1000 Years
Denisovan
26.39%
25.76%
32.70%
1.99%
120,000 BP
0.22%
Neanderthal
3.74%
2.79%
36.60%
3.27%
80,000 BP
0.047%
Heidelbergensis
4.27%
3.30%
37.61%
3.30%
430,000 BP
0.0099%
Ancient Egyptian
3.74%
0.17%
35.23%
8.32%
4,000 BP
.935%
Again, note that Denisovan, Neanderthal, and Heidelbergensis are all archaic humans. In contrast, the Ancient Egyptians are of course ancient, but not archaic. The dataset contains 664 rows, 76 of which are archaic or ancient, which leaves 588 rows for the comparisons produced above. As a result, even though the table above was produced using only 4 input genomes, the results were generated comparing each of the 4 input genomes to all 588 complete, modern human mtDNA genomes in the dataset. The plain implication is that modern human mtDNA is evolving faster than archaic human mtDNA, since, e.g., the Ancient Egyptian genome has an average total rate of change equal to that of the Neanderthals, despite having only 4,000 years to achieve this total change, in contrast to the roughly 120,000 years that have passed since the Neanderthal genome. Technically, we should only be testing genomes we believe to be descended from the archaic / ancient genomes, since e.g., it is theoretically possible that a modern person has mtDNA that predates the Ancient Egyptian genome, since mtDNA is so stable. That said, the bottom line is that this is a measure of the variability of a particular maternal line, and the amount of mutation cannot exceed that variability. For this and other reasons, more studies are required, but this is an interesting observation.
My research shows unequivocally, that archaic humans are still alive today, in that many living humans carry archaic mtDNA. The obvious question is, how did archaic humans survive for so long? The answer is, they probably didn’t, but their mtDNA did, just like the widely accepted fact that many living humans carry archaic DNA generally. What makes mtDNA unique, is that it is so stable, passed from a mother to its offspring, with basically no mutations at all, even over thousands of years. One estimate claims that one mutation occurs roughly every 7,990 years, though this estimate is qualified and plainly subject to doubt. I show below that assuming this is correct, Denisovan mtDNA existed about 38,000,000 years ago.
This is obviously way earlier than anyone thinks, but it’s not totally absurd, especially in light of relatively recent finds, including Graecopithecus, which was dated to 7.2 million years ago, in Greece, not Africa, which of course implies it’s possible the species emerged much earlier in Africa itself. Also note that we’re only discussing mtDNA, not the full genome. As a result, the claim is limited to the existence of Denisovan mtDNA, not the full genome. The discussion below of course considers the case that the estimate of 7,990 years per mutation is simply wrong, which is arguably the point of this note. Specifically, not all systems have stable averages over time, and a system as complex as the human genome of course might not behave in a predictable, stable manner.
Alignment, Insertions, and Deletions
Assume you have two copies of the exact same genome, and call them A and B. Note that mtDNA is N = 16,579 bases long, and as a result, the match count between genomes A and B is 16,579 bases, or 100% of the genome. Now insert a random base in genome B, at index 2. This will shift every base after the first index in B, by 1 position. This should cause the remaining N-1 bases to match to genome A about 25% of the time. That is, because we’ve shifted one of the otherwise identical genomes by one base, whatever bases that happen to match post insertion, should be the result of chance, and because there are four possible bases, the probability of a match is 1/4. Note that a deletion will cause an analogous reduction to chance. As a result, a single insertion or deletion will cause the match count to drop to around chance, after the index of the insertion or deletion.
The work I present in, “A New Model of Computational Genomics” [1], makes use of a global alignment, which means that when comparing two genomes, you assign each base an index, and the comparisons are made by testing whether the bases are equal at each index. The match count is simply the total number of matching bases. See [1] generally. In contrast, local alignments take segments from a given genome A (e.g., bases 1 through 100), and attempt to find the highest match count anywhere in genome B (e.g., bases 100 through 200). This would therefore, ignore insertions and deletions, since e.g., in the example above, a local alignment would search all of genome A for the best match, which would produce a match count of N (i.e., 100% of the genome), with one “gap” to account for the insertion. In contrast, a global alignment (i.e., just counting matching corresponding bases) would produce a match count of 1 + approximately 0.25*(N-1) (i.e., the first matching base, plus approximately 25% of the remaining N-1 bases).
Insertions and deletions are, at least anecdotally, very impactful in terms of the affect they have, since, e.g., Williams Syndrome, Down Syndrome, and many others, are caused by insertions and deletions. As a result, it’s not surprising that local alignments don’t seem terribly useful in terms of predictive power, because they effectively ignore insertions and deletions, creating very high match counts across all human mtDNA. In contrast, the software in [1], makes use a global alignment, which ultimately allows ethnicity to be predicted with approximately 80% accuracy.
Application to Data
As noted in [1], and many other research notes I’ve written, there are plenty of modern living humans with archaic mtDNA, in particular, Denisovan mtDNA. Denisovans test as the common ancestor of all archaic humans, suggesting that they are in fact the first humans. Though technically the modern people of Cameroon test as the ancestors of the Denisovans, which is again possible because mtDNA is so stable, I’ll work instead with the actual Denisovan genomes in my dataset, which were all taken from the NIH database. The goal of this section is to approximate the date of the first Denisovans, given the genomes of modern living humans that carry Denisovan mtDNA, and the actual Denisovan genomes recovered from Siberia. There are 8 such Denisovan genomes in the dataset, out of a total of 664 genomes. All genomes are complete mtDNA genomes, again taken from the NIH database.
If we fix a minimum match threshold of 50% of the genome, we find that 82 non-Denisovan genomes are at least a 50% match to at least one Denisovan genome. These are living, modern humans that carry Denisovan mtDNA. The average match count over all such genomes is 11,779.32 bases, or 71.05% of the full genome. This means that since the Denisovan cave, 100% – 71.05% = 28.95% of the genome has mutated. This is 4,799.62 bases.
Though the rate at which mtDNA mutates is still a subject of discussion, as noted above, one cited figure is one mutation per 7,990 years. This would put the age of the Siberian Denisovans at 38,348,963.80 years before the present. This is way out of the ballpark for the low-end of what I’ve seen regarding the dates of these finds, which is around 300,000 years ago. As noted above, it’s at least possible that the modern living Denisovans instead carry the mtDNA of the ancestors of the Siberian Denisovans, which would again force us to reject the date of 38,348,963.80 years before the present. However, the data suggests this is not the case. See Section 6 of [1] generally.
It could also be the case that a single insertion or deletion is causing the match count to drop to around 70% of the genome when comparing the Siberian Denisovans to modern living humans. That is, there’s a single insertion or deletion further down the genome that causes the balance of the genome match count to drop to around 70%. This would not require that much time, since it is technically a single mutation. We can however rule this out by looking at the distribution of the matching bases along the genome. This can be done by grouping sequential bases (i.e., bases 1 through K, K+1 through 2K, etc), and then counting the percentage of matching bases in those segments. If the matching percentage of bases in each segment is always significantly above 25%, then it simply cannot be the case that the resultant match count is due to a single insertion or deletion within a given segment. The chart below shows the average percentage of matching bases for all 8 of the Siberian Denisovan genomes when compared to all other genomes that have at least a 50% match, breaking the full genome into 100 segments of 165 bases each.
You can plainly see that it’s not the result of a single insertion or deletion, since the match count is always above 40% of the bases in each segment. That said, there is still plainly a portion of the genome from around segment 5 to segment 40, that seems to have been impacted by insertions and deletions, but this is distinct from a single trivial insertion or deletion. As a result, we have an enormous amount of change to account for when comparing Siberian Denisovan mtDNA to the mtDNA carried by some modern, living humans. This again implies that either the estimated rate of mutation is wrong (probably correct) or the dates associated with the Siberian cave are way off (not as convincing). The software for this is below, and the balance of the software can be found in [1].
In a previous article, I showed that the people of Cameroon test as the ancestors of Heidelbergensis, Neanderthals, and Denisovans, with respect to their mtDNA. The obvious question is, how is it that archaic humans are still alive today? The answer is that they’re probably not truly archaic humans, but that their mtDNA is truly archaic. This is possible for the simple reason that mtDNA is remarkably stable, and can last for thousands of years without changing much at all. However, there’s still the question of where modern humans come from, i.e., is there a group of people that test as the common ancestors of modern human populations. The answer is yes, and it’s the Phoenicians, in particular, a group of mtDNA genomes found in Puig des Molins. Astonishingly, the Phoenicians test as the common ancestor of the Pre-Roman Egyptians (perhaps not terribly astonishing), and the modern day Thai and Sri Lankans, the latter two being simply incredible, and perhaps requiring a reconsideration of purported history.
The overall test is straight forward, and cannot be argued with: Given genomes A, B, and C, if genome A is the ancestor of genomes B and C, then it must be the case that genomes A and B, and A and C, have more bases in common than genomes B and C. This is a relatively simple fact of mathematics, that you can find in my paper, A New Model of Computational Genomics [1], specifically, in footnote 16. However, you can appreciate the intuition right away: imagine two people tossing coins simultaneously, and writing down the outcomes. Whatever outcomes they have in common (e.g., both throwing heads), will be the result of chance. For the same reason, if you start with genome A, and you allow it to mutate over time, producing genomes B and C, whatever bases genomes B and C have in common will be the result of chance, and as such, they should both mutate away from genome A, rather than developing more bases in common with each other by chance. This will produce the inequalities |AB| > |BC| and |AC| > |BC|, where |AB| denotes the number of bases genomes A and B have in common.
For the same reason, if you count the number of matches between two populations at a fixed percentage of the genome, the match counts between populations A, B, and C, should satisfy the same inequalities, for the same reason. For example, fix the matching threshold to 30% of the full genome, and then count the number of genomes between populations A and B that are at least a 30% match or more to each other. Do the same for A and C, and B and C. However, you’ll have to normalize this to an [0,1] scale, otherwise your calculations will be skewed by population size. My software already does this, so there’s nothing to do on that front.
In this case, I’ve run several tests, all of which use the second population-level method described above. We begin by showing that the Phoenicians are the common ancestor of the modern day Sri Lankans and Sardinians. For this, set the minimum match count to 99.65% of the full genome size. This will produce a normalized score of 0.833 between the Phoenicians and Sri Lankans, and 0.800 between the Phoenicians and Sardinians. However, the score between the Sri Lankans and the Sardinians is 0.200, which plainly satisfies the inequality. This is consistent with the hypothesis that the Phoenician maternal line is the ancestor of both the modern day Sri Lankans and Sardinians. Setting the minimum match count to 88.01% of the genome, we find that the score between the Phoenicians and the Pre-Roman Egyptians is 0.500, and the score between the Phoenicians and the Sri Lankans is 1.000. The score between the Pre-Roman Egyptians and the Sri Lankans is instead 0.000, again satisfying the inequality. This is consistent with the hypothesis that the Phoenicians are the common ancestor of both the Pre-Roman Egyptians and the modern day Sri Lankans.
This seems peculiar, since the Phoenicians are Middle Eastern people, and the genomes in question are from Ibiza. However, the Phoenicians in particular were certainly sea-faring people, and moreover, civilization in the Middle East goes back to at least Ugarit, which could date as far back as 6,000 BC. Though not consistent with purported history, this at least leaves open the possibility that people from the Middle East traveled to South Asia. This might sound too ambitious for the time, but the Phoenicians made it to Ibiza from the Middle East, which is roughly the same distance as the Middle East to Sri Lanka, both of which are islands. Once you’re in South Asia, the rest of the region becomes accessible.
If this is true, then it shouldn’t be limited to Sri Lanka, and this is in fact the case. In particular, the Thai also test as the descendants of the Phoenicians, using the same analysis. Even more interesting, both the modern day Norwegians, Swedes, and Finns test as the descendants of the Thai, again using the same analysis. Putting it all together, it seems plausible that early Middle Eastern civilizations not only visited but settled South Asia, and that some of them came back, in particular to Egypt, and Scandinavia. This could explain why the Pre-Roman Egyptians are visibly Asian people, and further, why Thai-style architecture exists in early Scandinavia. Though the latter might sound totally implausible, it is important to note that some Thai and Norwegian people are nearly identical on the maternal line, with about 99.6% of the genome matching. Something has to explain that. Also note that the Sri Lankan maternal line was present throughout Europe around 33,000 BC. This suggests plainly that many Europeans, and the Classical World itself, descend from the Phoenicians. That somewhat remote populations also descend from them is not too surprising, in this context.
Further, there are alarming similarities between the Nordic religions and alphabet, and the Canaanite religions and alphabet, in particular, the gods El / Adon and Odin, with their sons, Baal and Baldur, respectively. Once you place greater emphasis on genetic history, over written history, this story sounds perfectly believable. Further still, if people migrated back from South Asia to the West, then this should again not be limited to Scandinavia, and this is in fact the case. Astonishingly, the Pre-Roman Egyptians test as the descendants of the Thai people, using the same analysis. Obviously the Pre-Roman Egyptians were not the first Africans, and in fact, everything suggests they’re South Asian, and for the same reason, none of this implies that modern day Scandinavians are the first Scandinavians, and instead, again, it looks like many Norwegians and Finns are instead, again, South Asian.
Finally, this is all consistent with the obvious fact that the most advanced civilizations in the world, i.e., the Classical World, are all proximate to the Middle East, suggesting that the genesis of true human intelligence, could have come from somewhere near Phoenicia.
Large leaps in evolution seem to require too much time to make sense. Consider the fact that about 500 bases separate human mtDNA from that of a gorilla or a chimp. That’s a small percentage of the approximately 16,000 bases that make up human mtDNA, but the number of sequences that are 500 bases in length is 4^ 500, which has approximately 300 digits. As a consequence, claiming that reproductive selection, i.e., the birth of some large number of children, that were then selected for fitness by their environment, is the driver of the change from ape to man, makes no sense, as there’s simply not enough time or offspring for that to be a credible theory, for even this small piece of its machinery, which is the evolution of mtDNA.
However, if we allow for evolution at the cellular level in the individual, over the lifetime of the individual, then it could explain how e.g., 500 extra bases end up added to the mtDNA of a gorilla, since there are trillions of cells in humans. That is, floating bases are added constantly, as insertions, in error, and when lethal, the cell in question dies off. However, if not lethal, and instead beneficial, this could occur throughout the body of the organism, causing the organism to evolve within its own lifetime, by e.g., changing its mtDNA through such a large scale, presumably beneficial insertion, like the one that divides apes from humanity.
This implies four corollaries:
1. It is far more likely that any such benefits will be passed on from the paternal line, since men constantly produce new semen. In contrast, women produce some fixed number of eggs by a particular age. As a result, men present more opportunities to pass down mutations of this type, if those mutations also impact their semen.
2. There must be some women who are capable of producing “new eggs” after a mutation, otherwise the mutation that caused gorilla mtDNA to evolve into human mtDNA, wouldn’t persist.
3. If you argue instead that such drastic mutations occur in the semen or the eggs, then you again have the problem of requiring too much time, since it would require a large number of offspring, that are then selected for lethal and non-lethal traits. This is the same argument we dismissed above. That is, the number of possible 500 base insertions is too large for this to be a credible theory. As a consequence, drastic mutations cannot be the result of reproductive selection, period, and require another explanation, for which cellular mutations within the individual seem a credible candidate.
4. If true, then it implies the astonishing possibility of evolution within the lifetime of an individual. This sounds far fetched, but cancer is a reality, and is a failure at the cellular level, that causes unchecked growth. The argument above implies something similar, but beneficial, that occurs during the lifetime of an individual, permeating its body, and thereby impacting its offspring.
In a previous note, I showed that the Denisovans appear to be the common ancestor of both Heidelbergensis and Neanderthals, in turn implying that they are the first humans. Since writing that note, I’ve expanded the dataset significantly, and it now includes the people of Cameroon. I noticed a while back that the people of Cameroon are plainly of Denisovan ancestry. Because it’s commonly accepted that humanity originated in Africa, the Cameroon are therefore a decent candidate for being related to the first humans.
It turns out, when you test Cameroon mtDNA, it seems like they’re not only related to the first humans, they are in fact the first humans, and test as the ancestors of the Denisovans, Heidelbergensis, and the Neanderthals. You might ask how it’s possible that archaic humans survived this long. The answer is, mtDNA is remarkably stable, and so while the people of Cameroon are almost certainly not a perfect match to the first humans, it seems their mtDNA could be really close, since they predate all the major categories of archaic humans with respect to their mtDNA.
The overall test is straight forward, and cannot be argued with: Given genomes A,B, and C, if genome A is the ancestor of genomes B and C, then it must be the case that genomes A and B, and A and C, have more bases in common than genomes B and C. This is a relatively simple fact of mathematics, that you can find in my paper, A New Model of Computational Genomics [1], specifically, in footnote 16. However, you can appreciate the intuition right away: imagine two people tossing coins simultaneously, and writing down the outcomes. Whatever outcomes they have in common (e.g., both throwing heads), will be the result of chance. For the same reason, if you start with genome A, and you allow it to mutate over time, producing genomes B and C, whatever bases genomes B and C have in common will be the result of chance, and as such, they should both mutate away from genome A, rather than developing more bases in common with each other by chance. This will produce the inequalities |AB| > |BC| and |AC| > |BC|, where |AB| denotes the number of bases genomes A and B have in common.
For the same reason, if you count the number of matches between two populations at a fixed percentage of the genome, the match counts between populations A, B, and C, should satisfy the same inequalities, for the same reason. For example, fix the matching threshold to 30%, and then count the number of genomes between populations A and B that are at least a 30% match or more to each other. Do the same for A and C, and B and C. However, you’ll have to normalize this to an [0,1] scale, otherwise your calculations will be skewed by population size. My software already does this, so there’s nothing to do on that front.
If it is the case that populations B and C evolved from population A, then the number of matches between A and B and A and C, should exceed the number of matches between B and C. The mathematics is not as obvious in this case, since you’re counting matching genomes, rather than matching bases, but the intuition is the same. Just imagine beginning with population A, and replicating it in populations B and C. In this initial state, the number of matching genomes between A and B, A and C, and B and C, are equal, since they’ve yet to mutate away from A (i.e., they are all literally the same population). As populations B and C mutate, the number of matching genomes between B and C should only go down as a function of time, since the contrary would require an increase in the number of matching bases between the various genomes, which is not possible at any appreciable scale. Again, see [1] for details.
In the first note linked to above, I show that the Denisovans are arguably the common ancestors of both Heidelbergensis and the Neanderthals. However, if you use the same code to test the Cameroon, you’ll find that they test as the common ancestor of the Denisovans, Heidelbergensis, and the Neanderthals. This is just not true of other populations that are related to Denisovans. For example, I tested the Kenyans, the Finns, and the Mongolians, all of which have living Denisovans in their populations (at least with respect to their mtDNA) and they all fail the inequalities. Now, there could be some other group of people that are even more archaic than the Cameroon, but the bottom line is, this result is perfectly consistent with the notion that humans originated in Africa, migrated to Asia, and then came back to both Europe and Africa, since e.g., about 10% of Kenyans are a 99% match to South Koreans and Hawaiians, and the Pre-Roman Ancient Egyptians were visibly Asian people, and about 40% of South Koreans are a 99% match to the Pre-Roman Ancient Egyptians.
The updated dataset that includes the Cameroons, and others, is available here. You’ll have to update the command line code in [1] to include the additional ethnicities, but it’s a simple copy / paste exercise, which you’ll have to do anyway to change the directories to match where you save the data on your machine.






{kind=link}