I came to the conclusion last night, that I may have wasted a lot of time, thinking about time as an actual dimension of space. In my defense, I’m certainly not the first physicist or philosopher to do so. Specifically, my entire paper, A Computational Mode of Time-Dilation [1], describes time as a measurement of physical change, not a dimension. Nonetheless, it produces the correct equations for time-dilation, without treating time as a dimension, though for convenience, in a few places, I do treat it as a dimension, since a debate on the corporeal nature of time is not the subject of the paper, and instead, the point is, you can have objective time, and still have time-dilation.
As a general matter, my view now is that reality is a three-dimensional canvas, that is updated according to the application of a rule, effectively creating a recursive function. See Section 1.4 of [1]. Because [1] is years old at this point, this is obviously not a “new” view, but one that I’ve returned to, after spending a lot of time thinking about time as an independent dimension, that could, e.g., store all possible states of the Universe. The quantum vacuum was one of the primary drivers for this view, specifically, that other realities temporarily cross over into ours, and because that’s presumably a random interaction, you should have a net-zero charge (i.e. equal representation from all charges), momentum, etc, on average, creating an otherwise invisible background to reality, save for extremely close inspection.
I don’t think I’m aware of any experiment that warrants such an exotic assumption, and I’m not even convinced the quantum vacuum is real. As such, I think it is instead rational to reject the idea of a space of time, until there is an experiment that, e.g., literally looks into the future, as opposed to predicting the future using computation.
I’ll concede the recursive function view of reality has some problems without time as a dimension, because it must be implemented in parallel, everywhere in space, otherwise, e.g., one system would update its states, whereas another wouldn’t, creating a single reality with multiple independent timelines. This is not true at our scale, and I don’t think there’s any experiment that shows it’s true at any scale. So if time doesn’t really exist as a dimension, we still need the notion of syncopation, which is in all fairness, typically rooted in time. But that doesn’t imply time is some form of memory of the past, or some projection of the future.
This is plainly an incomplete note, but the point is to reject the exotic assumptions that are floating around in modern physics, in favor of something that is far simpler, yet works. Reality as a recursive function makes perfect sense, taking the present moment, transforming it everywhere, producing the next moment, which will then be the present, with no record of the past, other than by inference from the present moment.
We’re still left with the peculiar fact that all of mathematics seems immutable (e.g., all theorems of combinatorics govern reality, in a manner that is more primary than physics, since they can never change or be wrong), but that doesn’t imply time is a dimension, and instead, my view, is that mathematics is beyond causation, and simply an aspect of the fabric of reality, whereas physics is a rule, that is applied to the substance contained in reality, specifically, energy. Physics doesn’t seem to change, but it could, in contrast, mathematics will never change, it’s just not possible.




 , it’s not clear how you can achieve an accuracy that is lower than
, it’s not clear how you can achieve an accuracy that is lower than  , as a vector
, as a vector 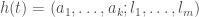 , where each
, where each 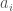 is the value of assets of type
is the value of assets of type  owned by the credit, and each
owned by the credit, and each  is the value of liabilities of type
is the value of liabilities of type 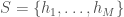 , that were sampled over time. That is,
, that were sampled over time. That is, 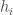 is actually a time-series of a given credit, and we can evaluate
is actually a time-series of a given credit, and we can evaluate  , for any
, for any 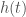 , which will produce a cluster of similar credits. Because our dataset contains time-series data for each of the credits returned in the cluster, we can form possible future paths for
, which will produce a cluster of similar credits. Because our dataset contains time-series data for each of the credits returned in the cluster, we can form possible future paths for 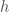 will look like, given its present state
will look like, given its present state  , we will construct a set of possible future paths for
, we will construct a set of possible future paths for 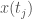 is in the cluster associated with
is in the cluster associated with  . We do this again, for all such
. We do this again, for all such  , and continue as desired, and this will produce a dataset of possible future paths for the credit, which will grow exponentially as a function of time, and at each ordinal interval of time, there will be some probability of default based upon the dataset.
, and continue as desired, and this will produce a dataset of possible future paths for the credit, which will grow exponentially as a function of time, and at each ordinal interval of time, there will be some probability of default based upon the dataset.
{kind=link}