I’ve put together a very simple model of genetic inheritance that mimics sexual reproduction. The basic idea is that parents meet randomly, and if they satisfy each other’s mating criteria, they have children that then participate in the same population. Individuals die at a given age, and the age of mortality is a function of genetic fitness, and overall it’s already pretty good, though I have planned improvements.
However, that’s not what’s important, though I will follow up tonight with more code, and instead, I stumbled upon what looks like a theorem of maximizing fitness through sexual reproduction. Specifically, if individuals mate by setting a minimum total fitness that is approximately the same as theirs (i.e., my mate is roughly as fit, overall, as I am), and that mate is at the same time maximally genetically distinct from the individual in question, then you maximize the fitness of the population. So to be crude, if I’m short, you’re tall, but neither of us are notably short or tall. As a consequence, we have roughly the same total fitness, but maximally different individual traits.
Just imagine for example two roughly unfit people, that are nonetheless maximally distinct from one another, in that if an aspect of one individual is minimally fit, the other is maximally fit, though neither are fit in total. Their children could inherit the best of their respective genomes, provided they have a sufficiently large number of children. As a consequence, there is at least a chance they have a child that is categorically more fit than either of them. Of course most of the outcomes will not be the ideal case where the child inherits the best of both parent’s genes, but compare this to two identically unfit people (i.e., incest between twins). In that case, the chance the child will be superior to the parents is much lower, and there is also a chance the child will be inferior. As a consequence, the case where the parents are maximally different, creates at least the possibility of superior offspring.
This will be true of all levels of fitness in a population, and as a consequence, a population that seeks out diverse mates, should be categorically superior to a population that seeks out identical mates, provided the environment kills off the weakest of all populations. Moreover, without the possibility of upside, you leave open the possibility that you fail to beat entropy, and as a result, attempting to maintain the status quo could lead to annihilation. That is, there’s definitely error in genetic reproduction, and if the upside of evolution doesn’t outstrip the downside of error, it’s at least plausible that the species just dies due to failure to evolve.
You can also think about this in terms of financial markets, specifically, by maximizing diversity between mating partners, but being reasonable with respect to total fitness, you increase the spread of outcomes, creating both upside and downside. If the environment kills the downside (which it definitely does in Nature), you leave the upside. In contrast, a homogenous strategy would be at best where it started, creating no upside, and leaving the downside for dead. If there’s error during reproduction, which there definitely is, then that alone could wipe out a homogenous strategy. The net point being, risk is necessary for survival, because without it, you don’t produce the upside that is necessary for survival to beat error, which is analogous to beating inflation. Continuing with this analogy, there’s a window of risk, within which, you get free genetic money, because Nature is so skewed against weakness, risk creates net returns within that window.
We can make this more precise by considering two genomes A and B, and assuming that alleles are selected from either A or B with equal probability, and corresponding to traits in the child of A and B. If we list the genes in order, along the genomes, we can assign each a rank of fitness at each index. Ideally, at a given index, we select the gene that has the higher of the two rankings (e.g., if A(i) > B(i), the child has the trait associated with A(i)). Because this process is assumed to be random, the probability of success equals the probability of failure, in that the better of the two genes is just as likely to be selected as the lesser of the two genes. As a consequence, there will be an exact symmetry in the distribution of outcomes, in that every pattern of successes and failures has a corresponding compliment. This is basically the binomial distribution, except we don’t care about the actual probabilities, just the symmetry, because the point again above is that lesser outcomes are subject to culling by the environment. Therefore, Nature should produce a resultant distribution that is skewed to the upside.
Note that this is in addition to the accepted view regarding the benefits of diversity, which is that diseases can be carried by mutated bases or genes, and because genetic diseases are often common to individual populations, mating outside your population reduces the risk of passing on the genetic diseases to children. It is instead, at least for humanity, a reproductive strategy that made sense a long time ago, when we were still subject to the violence of Nature. Today, diversity is instead more sensibly justified by the fact that it reduces the risk of inheriting genetic diseases. Note that wars are temporary in the context of human history, which is the result of hundreds of thousands of years of selection, and as such, mating strategies predicated upon the culling effect of Nature or other violence don’t really make sense anymore.
Here’s the code that as I noted is not done yet, but gets you there on intuition:
https://www.dropbox.com/s/1jp616fvcs2bxvx/Sexual%20Reproduction%20CMDNLINE.m?dl=0
https://www.dropbox.com/s/li0ilkvs8a3lmg0/update_mating.m?dl=0
https://www.dropbox.com/s/shzpgw85zv2uzla/update_mortality.m?dl=0



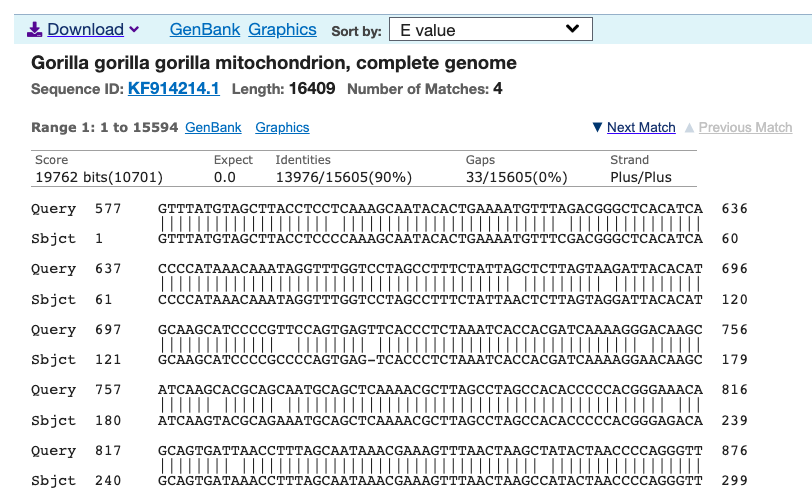
 , where
, where  is some set of indexes, where another genome
is some set of indexes, where another genome 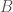 is included in the cluster if
is included in the cluster if  , you find that the average total number of matching bases between the full genome
, you find that the average total number of matching bases between the full genome 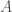 , and all such genomes
, and all such genomes  bases, incrementing by
bases, incrementing by  bases each iteration, and terminating at the full genome size of
bases each iteration, and terminating at the full genome size of  bases (i.e.,
bases (i.e.,  genomes in the dataset over each iteration. The random indexes are strictly superior, in that the average match count for every
genomes in the dataset over each iteration. The random indexes are strictly superior, in that the average match count for every 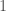 of the
of the 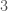 bases. As a consequence, if you concentrate the selected bases in a contiguous sequence, you’re creating overlap, since once you fix
bases. As a consequence, if you concentrate the selected bases in a contiguous sequence, you’re creating overlap, since once you fix 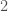 bases will likely be partially determined. Therefore, you maximize imputation by spreading the selected bases over the entire genome. Could be there an optimum distribution that isn’t random, yet not sequential? Perhaps, but the point is, random is not only good enough, but better than sequential, and therefore, the model presented in [1] makes perfect sense.
bases will likely be partially determined. Therefore, you maximize imputation by spreading the selected bases over the entire genome. Could be there an optimum distribution that isn’t random, yet not sequential? Perhaps, but the point is, random is not only good enough, but better than sequential, and therefore, the model presented in [1] makes perfect sense.
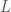 ,
,  , with a random starting index, and then also fixed
, with a random starting index, and then also fixed  . The random starting index for
. The random starting index for  , and counted how many genomes contained the sequence
, and counted how many genomes contained the sequence  . If random bases generate stronger imputation, then fewer genomes should contain the sequence
. If random bases generate stronger imputation, then fewer genomes should contain the sequence 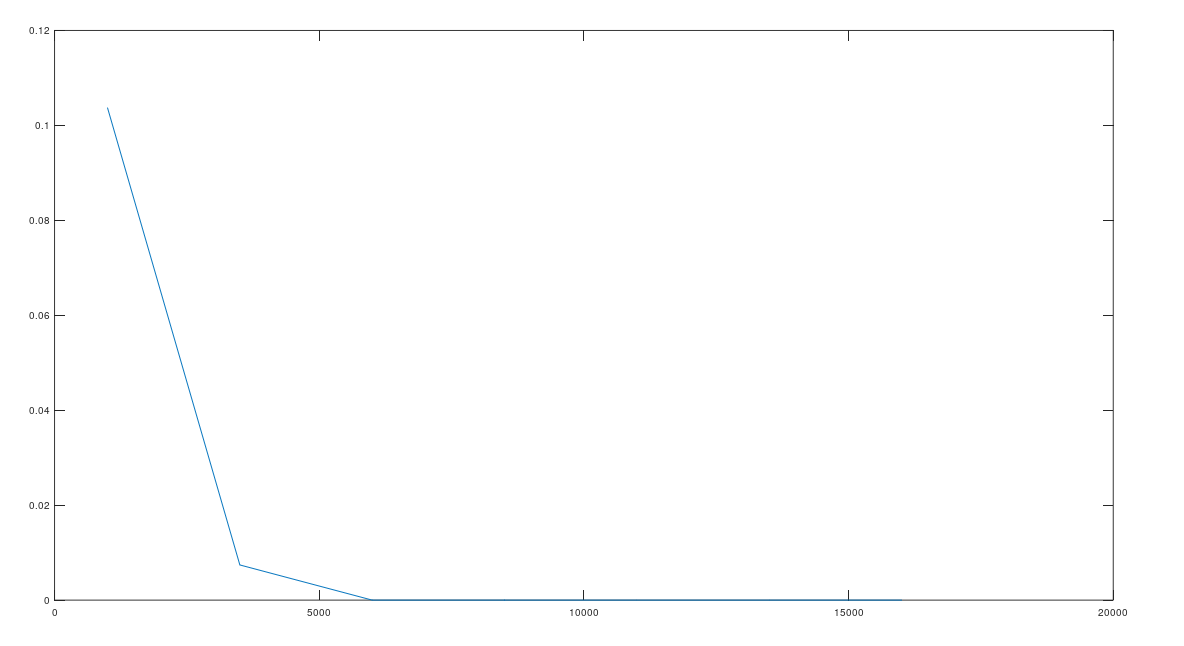

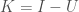 . In the case of a single observation of a given system, Information is assumed to be given by the maximum entropy of the system given its states, and so a system with
. In the case of a single observation of a given system, Information is assumed to be given by the maximum entropy of the system given its states, and so a system with 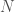 possible states has an Information of
possible states has an Information of  . The Uncertainty is instead given by the entropy of the distribution of states, which could of course be less than the maximum entropy given by
. The Uncertainty is instead given by the entropy of the distribution of states, which could of course be less than the maximum entropy given by  . If it turns out that
. If it turns out that  , then
, then  . All of this makes intuitive sense, since, e.g., a low entropy distribution carries very low Uncertainty, since it must have at least one high probability event, making the system at least somewhat predictable.
. All of this makes intuitive sense, since, e.g., a low entropy distribution carries very low Uncertainty, since it must have at least one high probability event, making the system at least somewhat predictable.