Yesterday I presented even more evidence that you get stronger imputation using random bases in a genome, as opposed to sequential bases in a genome. I already presented some evidence for this claim in my paper A New Model of Computational Genomics (see Section 7) [1]. Specifically, I showed in [1] that when calculating the nearest neighbor of a partial genome, you are more likely to map to the true nearest neighbor if you use random bases, as opposed to sequential bases. This suggests that imputation is stronger when using a random set of bases, as opposed to a sequential set of bases. The purpose of Section 7 was to show that using random bases is at least workable, because the model presented is predicated upon the assumption that you don’t need to look for genes or haplogroups to achieve imputation, so I didn’t really care whether or not random bases are strictly superior, though it seems that they are.
Specifically, if you build clusters using a partial genome 

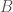

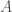




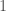
This might seem surprising, since so much of genetics is predicated upon genes and haplogroups, but it makes perfect sense, since, e.g., proteins are constructed using sequences of 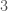
Here’s the dataset and the code:
https://www.dropbox.com/s/ht5g2rqg090himo/mtDNA.zip?dl=0
https://www.dropbox.com/s/9itnwc1ey92bg4o/Seq_versu_Random_Clusters_CMDNLINE.m?dl=0
Discover more from Information Overload
Subscribe to get the latest posts sent to your email.
