I’ve updated the dataset to account for ethnicity specifically, and so now all genomes have provenance directly tied to e.g., the Chinese, as opposed to simply being sampled in China, by a person that might not be Chinese. This applies to all of the 24 ethnicities, for a total of 341 complete mtDNA genomes. The overall number of rows increased, but some nationalities were reduced in size, because I wasn’t able to confirm ethnicity. The results really didn’t change much at all, so the last few articles I wrote still stand, but I’m still kicking the tires on all of this. That said, this dataset is now thoroughly diligenced, and again includes the raw genomes, together with links to the NIH Database for each genome included in the dataset.
One hypothesis I had that turned true, is that as you increase the number of rows, populations become increasingly concentrated in themselves. As a result, with a huge database like the NIH has, you can probably find a near perfect match for any two populations, but a truly perfect match seems to be limited in number for remote groups. As a consequence, working with a smaller dataset makes sense, if you want to uncover interesting relationships between populations. That said, running a BLAST search is a good sanity check for any hypothesis.


One shocker, I found a 4,000 year old, Pre-Roman Egyptian complete mtDNA genome, and its closest matches in the dataset below are a Norwegian and a Dane (graph above). I just proofed it using BLAST, and it seems like it’s legit, as a near perfect match comes up with an ethnic Norwegian. This is consistent with the last few posts I’ve shared, showing a strong genetic connection between the Nordic people (if you include the Scotts) and a global population that reaches all the way from South America to Polynesia, that seems to predate the Vikings. I would wager again, the world was globalized, a really long time ago, and it could have been the result of sailboats and possibly early telescopes, or something close that would allow you to spot land over huge distances, rather than meander at sea and therefore almost certainly die in places like Polynesia, where the distances between islands are way beyond human vision.
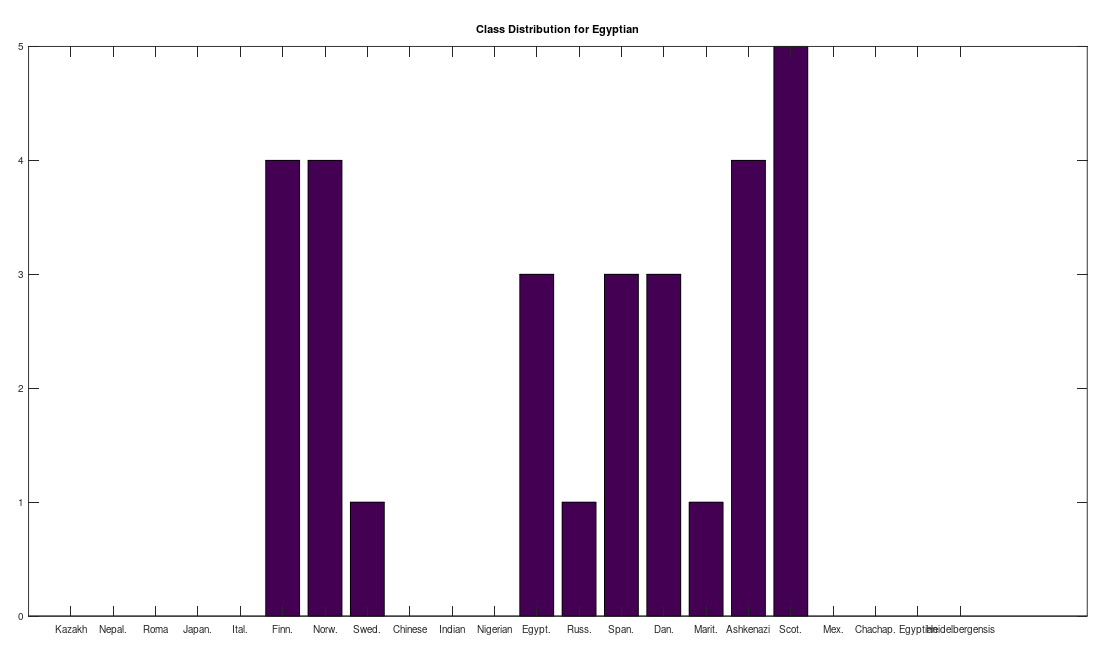
I tested it even further, and bizarrely, the Scotts yet again, show up as the dominant group for the 4,000 year old ancient Egyptian genome. Note again, this is a dataset of complete genomes, including two African countries (Nigeria and Egypt itself), and the Scotts are the dominant group when you set the minimum matching base count to 99.7% of the genome. This is not, to my knowledge, consistent with known history, and suggests yet again, the world was significantly globalized, a very long time ago. Interpreted literally, this means 5 out of the 20 Scottish genomes in the dataset were a 99.7% match to a 4,000 year old Egyptian genome.
The bottom line conclusion is that many Scottish people are plainly of Ancient Egyptian heritage on their maternal line. You can fuss that the number of genomes per population is not uniform, but this doesn’t change the percentage of Scotts that match, which is plainly high. Moreover, there are 20 modern Egyptian genomes and 20 Scottish genomes, and the Scotts plainly fit better. Further, there are 19, 20, and 18 Finnish, Norwegian, and Swedish genomes, respectively, and the Swedes are plainly not as closely related to the Ancient Egyptian genome, suggesting that it’s not a simple matter of geography. Finally, that any of the Northern Europeans are this closely related is simply baffling, since, e.g., why aren’t the Nigerians and Italians related? They’re geographically proximate, with some kind of sensible historical connection. This is irrefutable, and simply not consistent with known history.
Rather than give the Scotts any special credit, I think the takeaway is instead that the world was already diverse a very long time ago, and this is consistent with the distribution of aesthetics in the world before Christ, in particular in Ancient Egypt, which plainly depicts racially diverse people, though this is not true during Cleopatra’s reign, when people seemed more or less Mediterranean. On the left is the Berlin Green Head, in the center is Menkaure and Queen Khamerernebty II, and on the right is Nefertiti, images courtesy of Wikipedia, MFA Boston, and Wikipedia, respectively.



You’ll note that none of these people look Mediterranean, and in my opinion, they look to be of mixed heritage, demonstrating African, Asian, and European features. They could instead be so ancient that they are like the Khoisan, who have similar mixed features, but that is apparently not supported by the dataset, suggesting that they really were multi-racial people. When you look at the level of skill in their work, I have no trouble believing it, as they were plainly an advanced people, and it requires advanced people to produce multi-racial people, in particular, royalty. I doubt there are many living artists capable of producing works on this level, and regrettably, many people still struggle with simply getting along with superficially different people. The cruel justice of this work is that it’s all nonsense, and our histories have apparently been mixed up for quite a long time.
Here’s the dataset:











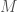 out of a total of
out of a total of 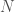 bases, you will produce better results if you randomly select the indexes of the bases, rather than use a contiguous sequence of bases of length
bases, you will produce better results if you randomly select the indexes of the bases, rather than use a contiguous sequence of bases of length 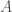 , and set of base indexes
, and set of base indexes  , knowledge of the bases
, knowledge of the bases  implies knowledge about the genome
implies knowledge about the genome 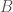 is the true nearest neighbor of
is the true nearest neighbor of 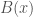 (i.e., the nearest neighbor of
(i.e., the nearest neighbor of 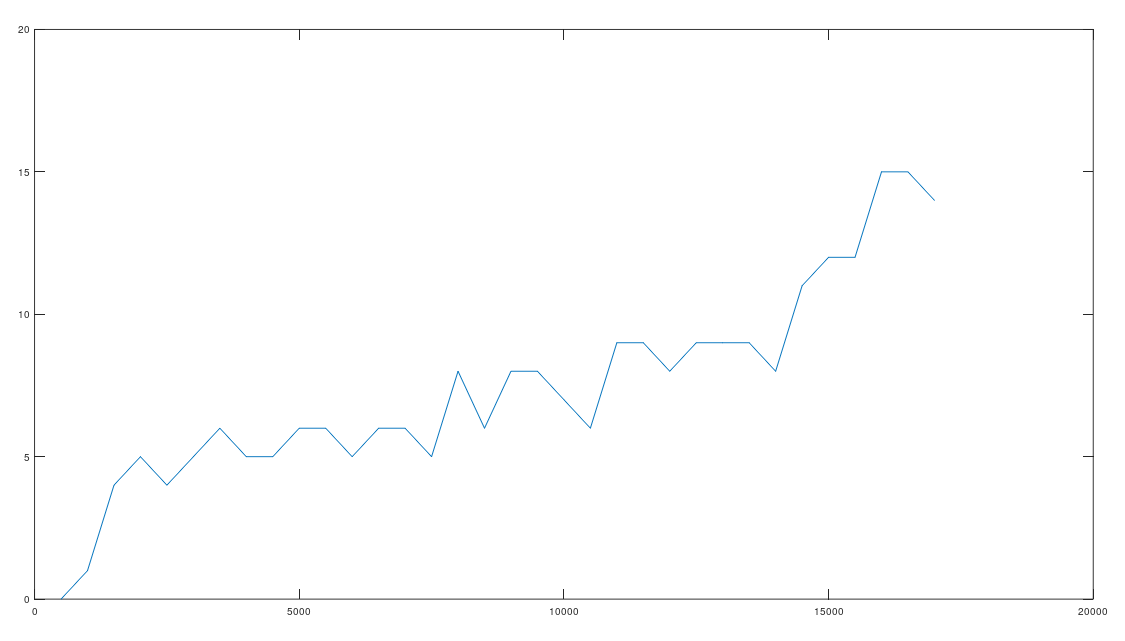
 . All of this can be understood by noting that if this were the result of chance, then once the nearest neighbor of a given row is fixed, the row that maximizes the match count beyond a given
. All of this can be understood by noting that if this were the result of chance, then once the nearest neighbor of a given row is fixed, the row that maximizes the match count beyond a given  ways. Note that a single successful outcome has a probability of about
ways. Note that a single successful outcome has a probability of about  , whereas
, whereas 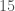 successful outcomes has a probability around
successful outcomes has a probability around  . As a consequence, it is not credible to claim that the graph above is the result of chance. Instead, it is more sensible to assume that imputation is a phenomenon justified beyond the existence of genes and haplogroups, and is instead a fundamental statistical property of genomes.
. As a consequence, it is not credible to claim that the graph above is the result of chance. Instead, it is more sensible to assume that imputation is a phenomenon justified beyond the existence of genes and haplogroups, and is instead a fundamental statistical property of genomes. . As such, when comparing two genomes and counting matching bases, after adjusting alignment, we are engaging in a series of
. As such, when comparing two genomes and counting matching bases, after adjusting alignment, we are engaging in a series of  matching bases over two genomes of length
matching bases over two genomes of length  , where
, where  bases. This is a number that has approximately
bases. This is a number that has approximately  digits, and cannot be calculated using an ordinary computer. The expected number of matching bases is
digits, and cannot be calculated using an ordinary computer. The expected number of matching bases is  bases. The standard deviation is
bases. The standard deviation is  . Note that the expected number of matching bases, and the standard deviation have nothing to do with observation, and are instead implied by the fact that comparing
. Note that the expected number of matching bases, and the standard deviation have nothing to do with observation, and are instead implied by the fact that comparing  .
. , which is at times higher than
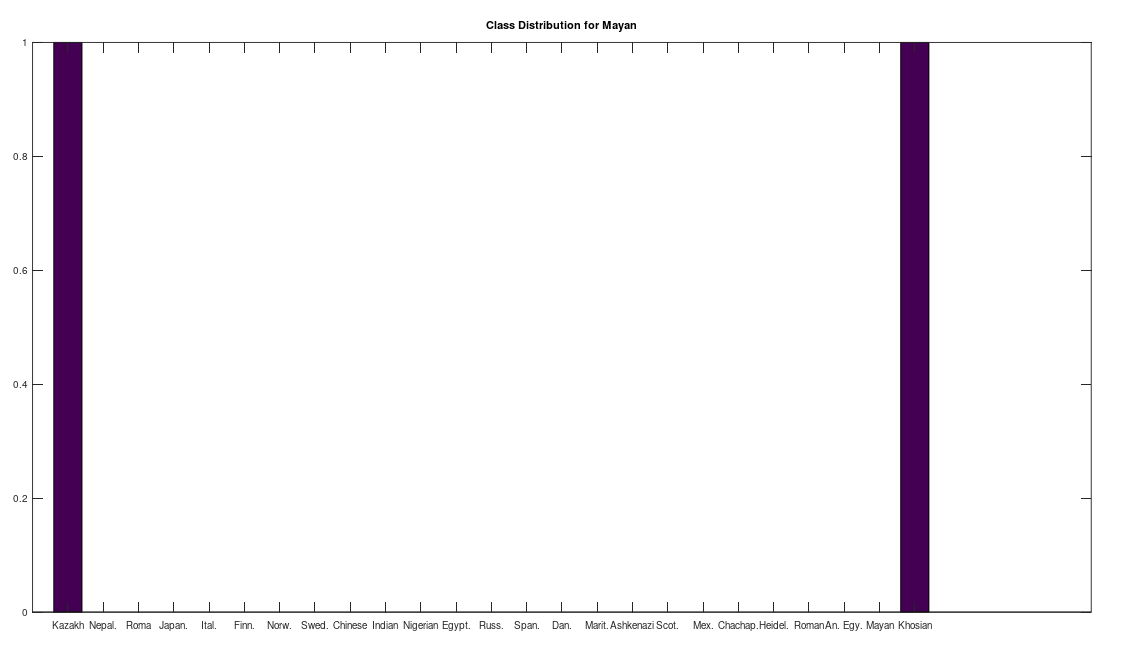
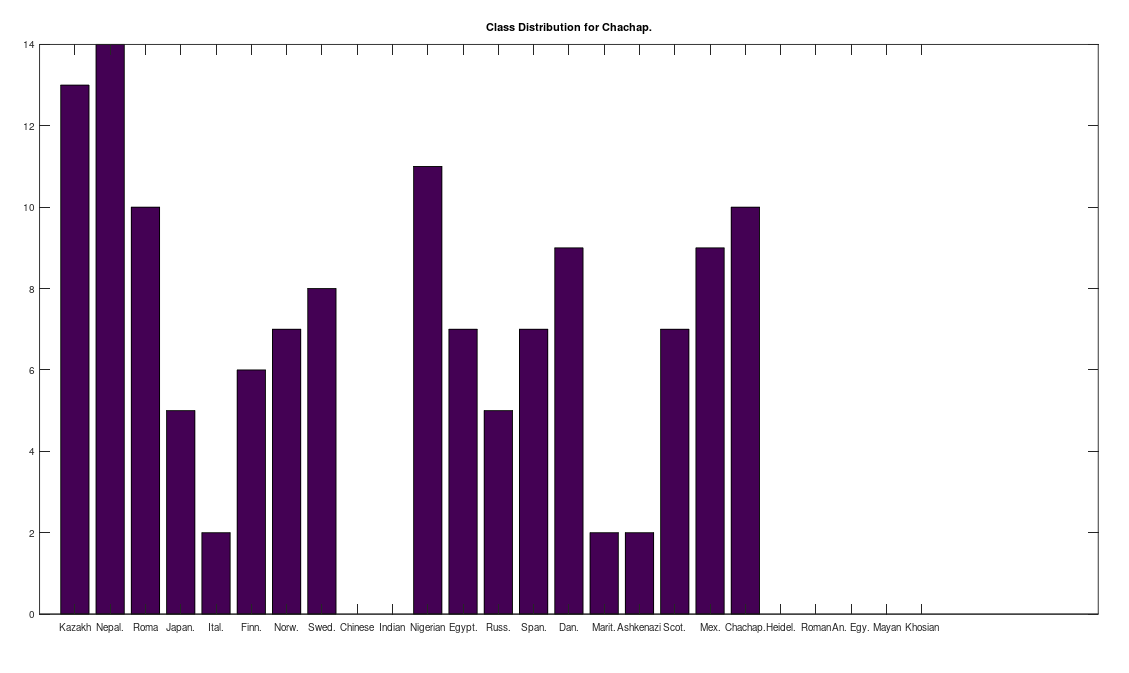
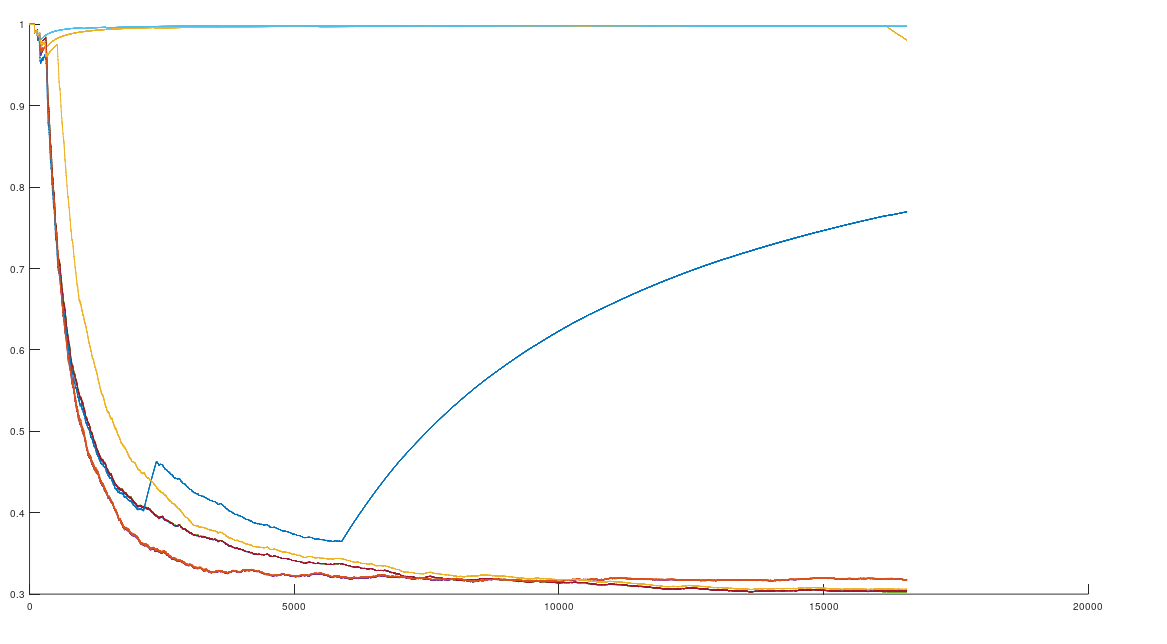
, which is at times higher than  , since many genomes contain missing entries, which do not contribute to the threshold. Using this threshold produces the following three distributions for the Maritime Archaic, Mayan, and Chachapoya, respectively, in that order below. Keep in mind, you simply cannot argue with these results, since mtDNA does not change much from one generation to the next, and as noted above, the notion that matches on this scale are the product of chance is simply not credible, and so these groups are genetically related as set forth below. The vertical axis counts the number of times a given population matched to the population in question, and the horizontal axis gives the names of the individual populations.
, since many genomes contain missing entries, which do not contribute to the threshold. Using this threshold produces the following three distributions for the Maritime Archaic, Mayan, and Chachapoya, respectively, in that order below. Keep in mind, you simply cannot argue with these results, since mtDNA does not change much from one generation to the next, and as noted above, the notion that matches on this scale are the product of chance is simply not credible, and so these groups are genetically related as set forth below. The vertical axis counts the number of times a given population matched to the population in question, and the horizontal axis gives the names of the individual populations.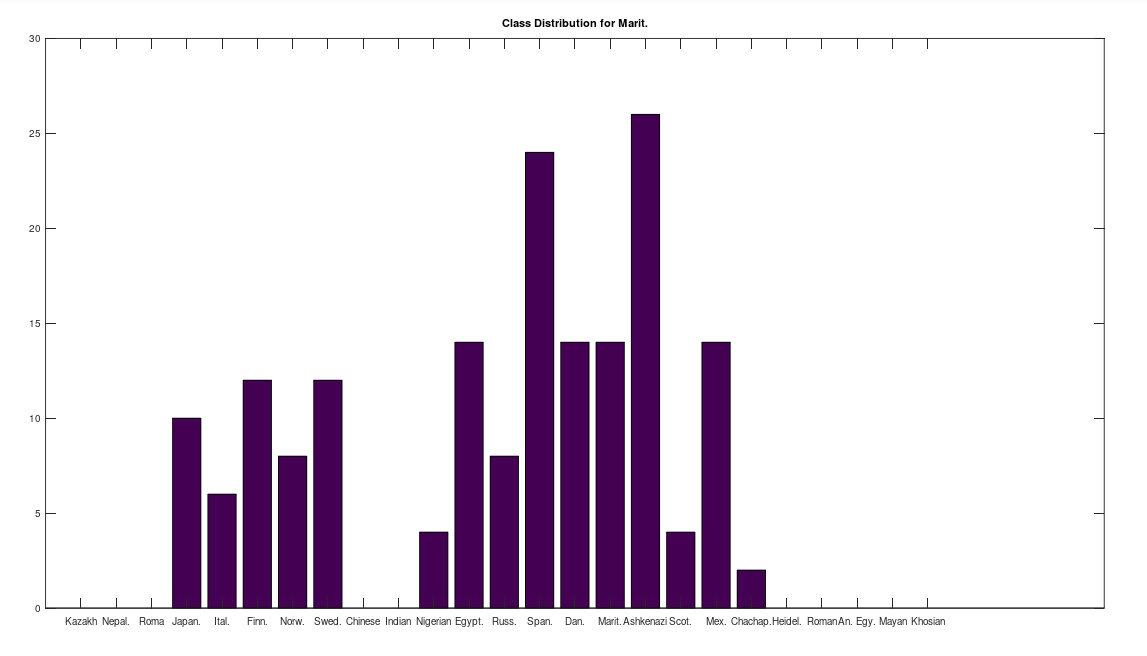
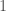 to
to  , thereby testing multiple independent starting indexes. If the sequence pushed past the end (i.e.,
, thereby testing multiple independent starting indexes. If the sequence pushed past the end (i.e.,  ), I simply started again from other side using modular indexes (i.e., modulo
), I simply started again from other side using modular indexes (i.e., modulo  to map to
to map to 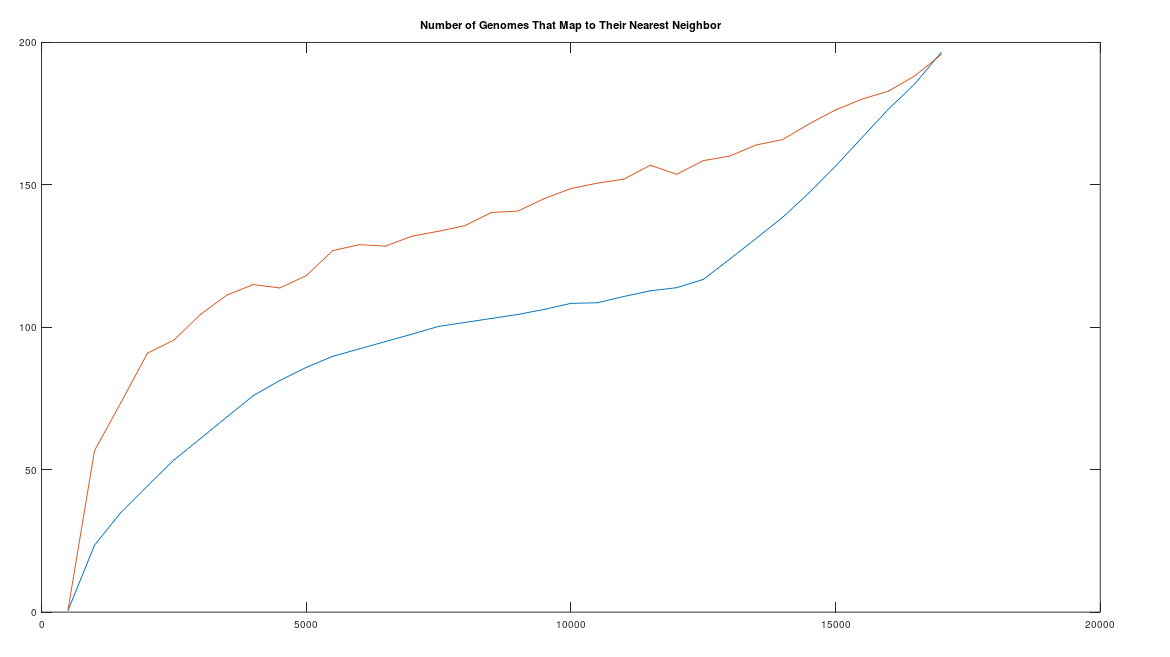




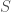 that produces signals over time, and assume that you record the signals generated. If
that produces signals over time, and assume that you record the signals generated. If  . The probability of producing two sequential observations is
. The probability of producing two sequential observations is  . The probability of producing two unequal observations is instead
. The probability of producing two unequal observations is instead  . As a consequence, it is more likely than not that the first two observations present two novel observations. Now assume instead that
. As a consequence, it is more likely than not that the first two observations present two novel observations. Now assume instead that  , with the probability of
, with the probability of  . This then implies that the probability of two novel observations is given by
. This then implies that the probability of two novel observations is given by  , whereas the probability of sequential
, whereas the probability of sequential 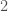 ‘s is given by
‘s is given by  . As is evident, the higher the entropy of a distribution, the greater the likelihood of novelty, though I’ll concede this is not a formal proof.
. As is evident, the higher the entropy of a distribution, the greater the likelihood of novelty, though I’ll concede this is not a formal proof.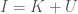 , where I would be in this case the maximum entropy of a source, and
, where I would be in this case the maximum entropy of a source, and 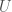 is its entropy, leaving Knowledge as the balance between the two. Applied in this case, a low entropy system provides some knowledge about its history, whereas a high entropy system does not.
is its entropy, leaving Knowledge as the balance between the two. Applied in this case, a low entropy system provides some knowledge about its history, whereas a high entropy system does not.