Begin with a sequence of DNA base pairs for each individual in a population of size 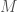 . Then separate these sequences into single strands of DNA. Assume that each DNA strand contains
. Then separate these sequences into single strands of DNA. Assume that each DNA strand contains 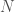 individual bases over
individual bases over  . Now construct a matrix
. Now construct a matrix 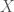 where each column of contains characters that define the DNA sequence of exactly one individual from the population. It follows that is a matrix with rows (i.e., the number of bases) and columns (i.e., the number of individuals in the population). You can think of the matrix as readable from the first row of a given column, down to the bottom row of that same column, which would define the genetic sequence for a given individual in the population.
where each column of contains characters that define the DNA sequence of exactly one individual from the population. It follows that is a matrix with rows (i.e., the number of bases) and columns (i.e., the number of individuals in the population). You can think of the matrix as readable from the first row of a given column, down to the bottom row of that same column, which would define the genetic sequence for a given individual in the population.
Now construct a new matrix  such that
such that  , where
, where 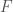 maps
maps 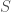 to
to  . That is, maps each of the four possible bases to the numbers 1 through 4, respectively. All we’ve done is encode the bases from the original matrix using numbers. Now for each row of , calculate the density of each of the four bases in the row, and store those four resultant densities in a matrix
. That is, maps each of the four possible bases to the numbers 1 through 4, respectively. All we’ve done is encode the bases from the original matrix using numbers. Now for each row of , calculate the density of each of the four bases in the row, and store those four resultant densities in a matrix 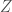 , that has rows and
, that has rows and  columns. That is, each of the four possible bases will have some density in each row of , which is then stored in a corresponding row of , with each of the four columns of containing the densities of
columns. That is, each of the four possible bases will have some density in each row of , which is then stored in a corresponding row of , with each of the four columns of containing the densities of  , respectively, for a given row of .
, respectively, for a given row of .
Further, construct a vector  with a dimension of
with a dimension of  , where row
, where row  of
of  contains the maximum entry of row of . That is, for every row of , which contains the densities for each of the four bases over every row, we find the base that is most dense for a given row, and store that density in the corresponding row of a new vector . Then, construct a binary vector
contains the maximum entry of row of . That is, for every row of , which contains the densities for each of the four bases over every row, we find the base that is most dense for a given row, and store that density in the corresponding row of a new vector . Then, construct a binary vector  that maps every element of to either
that maps every element of to either 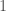 or
or 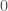 , depending upon whether or not the entry in question is greater than some threshold in
, depending upon whether or not the entry in question is greater than some threshold in ![[0,1]](https://s0.wp.com/latex.php?latex=%5B0%2C1%5D&bg=ffffff&fg=444444&s=0&c=20201002) . The threshold allows us to say that if e.g., the density of A in a given row exceeds
. The threshold allows us to say that if e.g., the density of A in a given row exceeds  , then we treat it as homogenous, and uniformly A, disregarding the balance of the entries that are not A’s. It follows that the longest sequence of consecutive
, then we treat it as homogenous, and uniformly A, disregarding the balance of the entries that are not A’s. It follows that the longest sequence of consecutive  in , is the longest sequence of bases common to the entire population in question (subject to the threshold). All of these operations have in the worst case a linear runtime, or less when run in parallel. As a consequence, we can identify DNA sequences common to an entire population in worst case linear time.
in , is the longest sequence of bases common to the entire population in question (subject to the threshold). All of these operations have in the worst case a linear runtime, or less when run in parallel. As a consequence, we can identify DNA sequences common to an entire population in worst case linear time.
The first step of mapping the bases to letters can be done independent of one another, and so this step has a constant runtime in parallel. The second step of calculating the densities can be accomplished in worst case linear time, for each row, since a sum over operands can be done with at most operations. The densities for each row can be calculated in parallel, and so this step requires a linear number of steps as a function of . The next step of taking the maximum densities can again be done in parallel for each row, and requires operations for a given row. The next step of comparing each maximum density for each row to a fixed threshold can again be done in parallel, and therefore requires constant time. The final step of finding the longest sequence requires exactly operations. As a result, the total runtime of this algorithm, when run in parallel, is  .
.
Attached is Octave code that implements this, and finds the longest genetic sequence common to a population (using the threshold concept). This can be modified trivially to find the next longest, and so on. This will likely allow for e.g., long sequences common to people with genetic diseases to be found, thereby allowing for at least the possibility of therapies.
Discover more from Information Overload
Subscribe to get the latest posts sent to your email.
